法尼基轉移酶及其抑制劑在腫瘤治療中的研究進展
2021-08-10 00:33:56胡清源蘇慧玲徐天瑞
中國藥理學通報 2021年8期
馮 辰,胡清源,田 銣,蘇慧玲,安 輸,徐天瑞
(昆明理工大學生命科學與技術學院,云南省高校靶點藥物篩選與利用重點實驗室,云南 昆明 650500)
惡性腫瘤是對人類危害最大、最為重要的致死性疾病,其發病率和死亡率均位于各類疾病前列[1]。針對惡性腫瘤的藥物治療策略也較多,如有各種特異性靶向關鍵受體和關鍵激酶等的抑制劑,它們可抑制誘發腫瘤發生發展關鍵信號通路的活化。其中Ras-MAPK (mitogen-activated protein kinase,MAPK) 信號通路和PI3K (phosphatidylinositol 3-kinase,PI3K)-Akt信號通路在腫瘤發生發展中的作用被研究的最為透徹,針對這兩條信號通路的抗腫瘤靶向藥物在臨床上也被應用的最多[2]。
自上個世紀60年代Ras原癌基因被發現以來,眾多的研究表明,Ras蛋白與腫瘤的發生發展密切相關,大約有1/3的人類腫瘤是因Ras基因突變而導致的[3]。但因Ras蛋白結構的獨特性,致使其缺乏藥物分子結合口袋,并使之難以成為小分子化合物作用的靶點,所以學界普遍認為Ras蛋白是“無藥可靶向的” (undruggable)[4]。所以,對Ras蛋白功能具有重要調控作用的FTase逐漸成為抗腫瘤藥物研發的焦點[5]。
FTase是一種膜結合蛋白,研究表明,其在腫瘤細胞的增殖、侵襲和代謝等過程中發揮著重要作用。FTase最主要的功能是其可通過催化作用,將一個含有15個碳原子的法尼基脂共價連接到靶蛋白羧基端四肽基序中的半胱氨酸巰基上,實現對靶蛋白的法尼基化和功能調控[6]。其中Ras是FTase最為重要的底物蛋白之一,只有當Ras蛋白被法尼基化后,其才能通過法尼基脂結合于細胞膜而被激活[7-8]。
1 Ras蛋白概述
Ras蛋白是一種細胞膜結合蛋白,它屬于GTPase家族成員,是一種小G蛋白。Ras蛋白家族含有H-Ras、N-Ras、K-Ras 4A和K-Ras 4B等4個成員,它們分別由H-Ras、N-Ras和K-Ras基因編碼生成,其中K-Ras 4A和K-Ras 4B是K-Ras基因的不同剪接體[9]。如前文所述,Ras基因突變是誘發大約1/3人類腫瘤的主要因素,而排在前三位致死率最高的腫瘤(肺腺癌、結直腸癌和胰腺癌)中,K-Ras的突變最為常見,分別在86%的胰腺癌組織、41%的結直腸癌組織和32%的肺腺癌組織中均檢測到K-Ras基因突變。K-Ras基因突變主要發生在第12位密碼子,該密碼子編碼甘氨酸 (G)。在肺癌組織中,第12位甘氨酸常常突變成半胱氨酸 (G12C),而在結直腸癌和胰腺癌組織中,甘氨酸常常突變成天冬氨酸 (G12D)[3,10]。
在正常細胞中,位于細胞膜的Ras蛋白可被表皮生長因子受體(epidermal growth factor receptor,EGFR)等受體及鳥嘌呤核苷酸交換因子(guanine nucleotide exchange factor,GEF)所激活。GEF可實現Ras蛋白由非活化狀態GDP-Ras向活化狀態GTP-Ras轉變,從而激活Ras蛋白及其下游MAPK和PI3K-Akt這兩條細胞主干信號通路,以維持細胞的正常增殖和分化等生理功能。GTP酶活化蛋白 (GTPase-activating protein,GAP)可催化GTP-Ras中GTP的水解,使Ras蛋白轉變成GDP-Ras狀態,反饋性地抑制Ras活性,防止Ras下游信號通路的持續激活[11]。而當Ras基因發生致癌突變時,GAP將無法水解GTP-Ras中的GTP,致使Ras一直處于激活狀態,從而導致Ras下游信號通路的持續異常活化及細胞的癌性轉變[11]。
2 FTase對Ras蛋白功能的調控及其在腫瘤發生發展中的作用
作為一種在胞質中剛被表達的Ras前體蛋白是沒有活性的,它需要經歷諸多翻譯后修飾包括聚異戊二烯化 (3個異戊二烯即可形成法尼基)、蛋白水解和羧基甲基化等才能轉變成有活性的Ras蛋白[12]。Ras蛋白的法尼基化是其翻譯后修飾的第一步。在FTase催化作用下,其可將一個含有15個碳原子的法尼基異戊二烯脂連接到Ras蛋白羧基端CAAX四肽基序的半胱氨酸巰基上,其中C是半胱氨酸,A是一種脂肪族氨基酸,X是絲氨酸或甲硫氨酸。Ras蛋白被法尼基化后,法尼基異戊二烯可以將Ras固定在內質網上。位于內質網上的Ras蛋白羧基端CAAX四肽基序中的AAX進而被RCE1(Ras-converting CAAX endopeptidase 1, RCE1)所酶切水解。之后在ICMT (isoprenylcysteine carboxyl methyltransferase)酶的作用下,一個羧甲基被連接到羧基末端的半胱氨酸上。最后,在棕櫚酰轉移酶 (palmitoyltransferase) 的作用下,兩個棕櫚酰長鏈脂肪酸基團被分別連接到羧甲基化半胱氨酸上游的兩個半胱氨酸上。此時,棕櫚酰化的Ras蛋白擁有一個較強的疏水性尾巴,該尾巴使Ras蛋白對細胞膜具有較高的親和力,并可將Ras蛋白固定于細胞膜,從而有利于Ras蛋白的激活[7]。
研究表明,FTase在肝癌、肺癌、卵巢癌和食管癌等癌組織中呈高表達態勢,其可通過促進腫瘤細胞增殖、侵襲、轉移、腫瘤血管生成和抑制腫瘤細胞凋亡而促進腫瘤的發生發展[6,13-14]。FTase可以使Ras、CENP-E (centromere-associated protein E,CENP-E)、CENP-F和spindly等蛋白法尼基化,上述蛋白又是細胞增殖和細胞周期進程的重要調控分子。FTase促進腫瘤發生發展的分子機制主要與Ras蛋白有關,因Ras-MAPK和Ras-PI3K-Akt信號通路異常活化在腫瘤細胞增殖、侵襲、轉移和血管生成過程中發揮著重要作用[14-15]。
3 FTase抑制劑
自從發現Ras蛋白的法尼基化對其功能至關重要以來,FTase抑制劑(FTIs) 逐漸成了抗腫瘤藥物研究熱點。根據FTIs結構及其抑制FTase的作用機制,可將FTIs分為4類:(1)CAAX四肽基序及其模擬物,(2)法尼基焦磷酸(Farnesyl pyrophosphate,FPP,它是法尼基的活化形式)模擬物,(3)過渡態模擬物,(4)其它化合物[16]。
最初的FTIs即為野生型K-Ras羧基端CVIM四肽,后續研究工作對該四肽化合物進行了改造,主要目的是為了減少該類FTIs骨架中的酰胺鍵,降低FTIs在體內被蛋白酶水解的風險,提高FTIs的半衰期。此外,還可通過替換CAAX四肽中的氨基酸A,以提高其活性。如將CVIM四肽化合物中異亮氨酸(I)替換成疏水性更強的苯丙氨酸(F),則CVFM四肽化合物的FTIs活性明顯增強[17](Fig 3)。
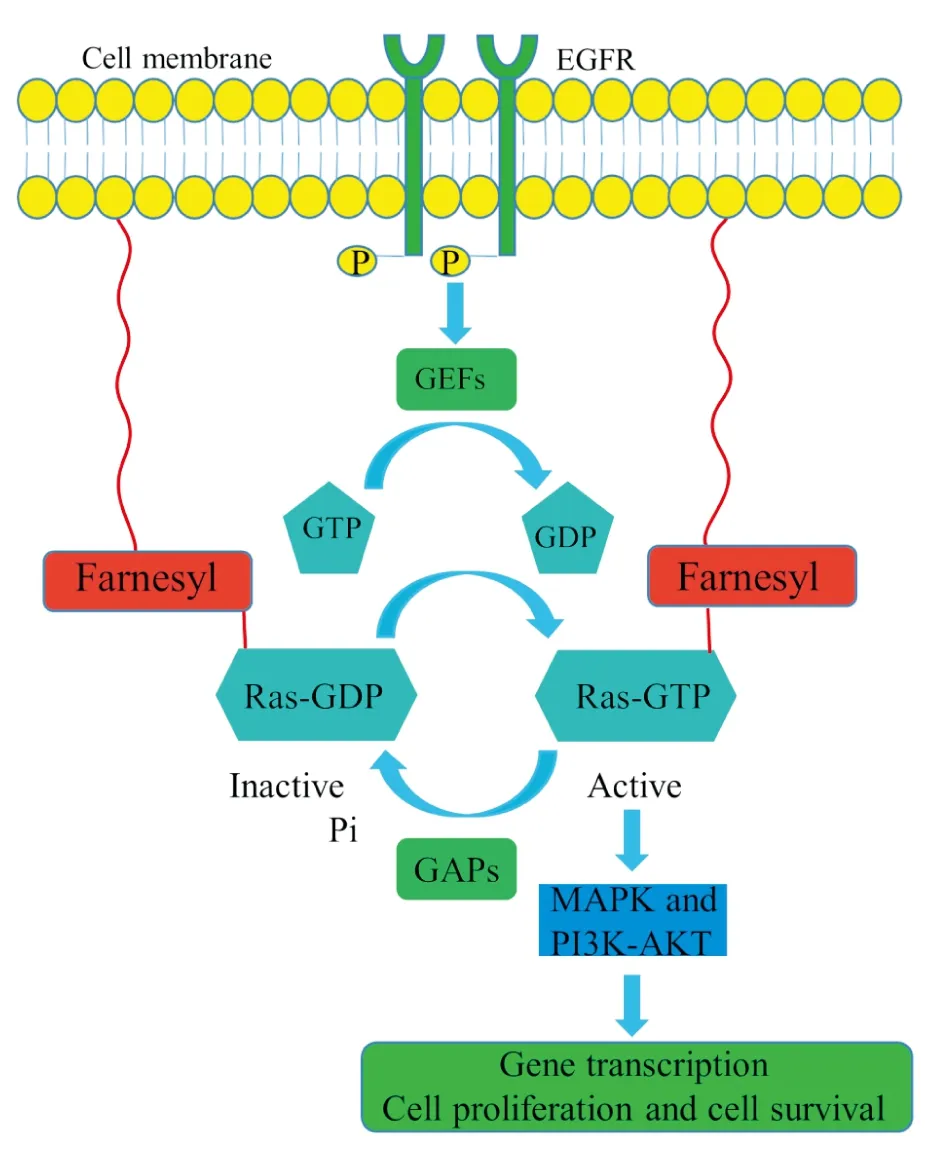
Fig 1 Schematic diagram of activation of Ras protein and its downstream MAPK and PI3K-Akt signaling pathways
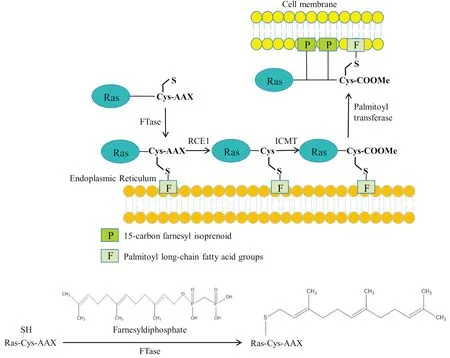
Fig 2 The farnesylation and palmitoylation modification of Ras protein
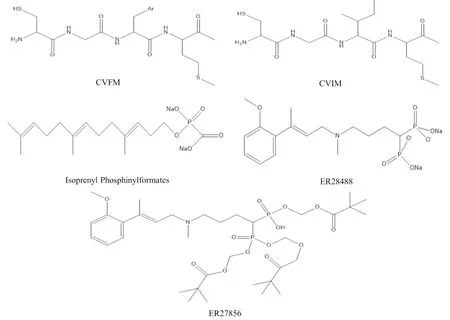
Fig 3 The chemical structures of CVFM, CVIM, isoprenyl phosphinylformates, ER28488 and ER27856
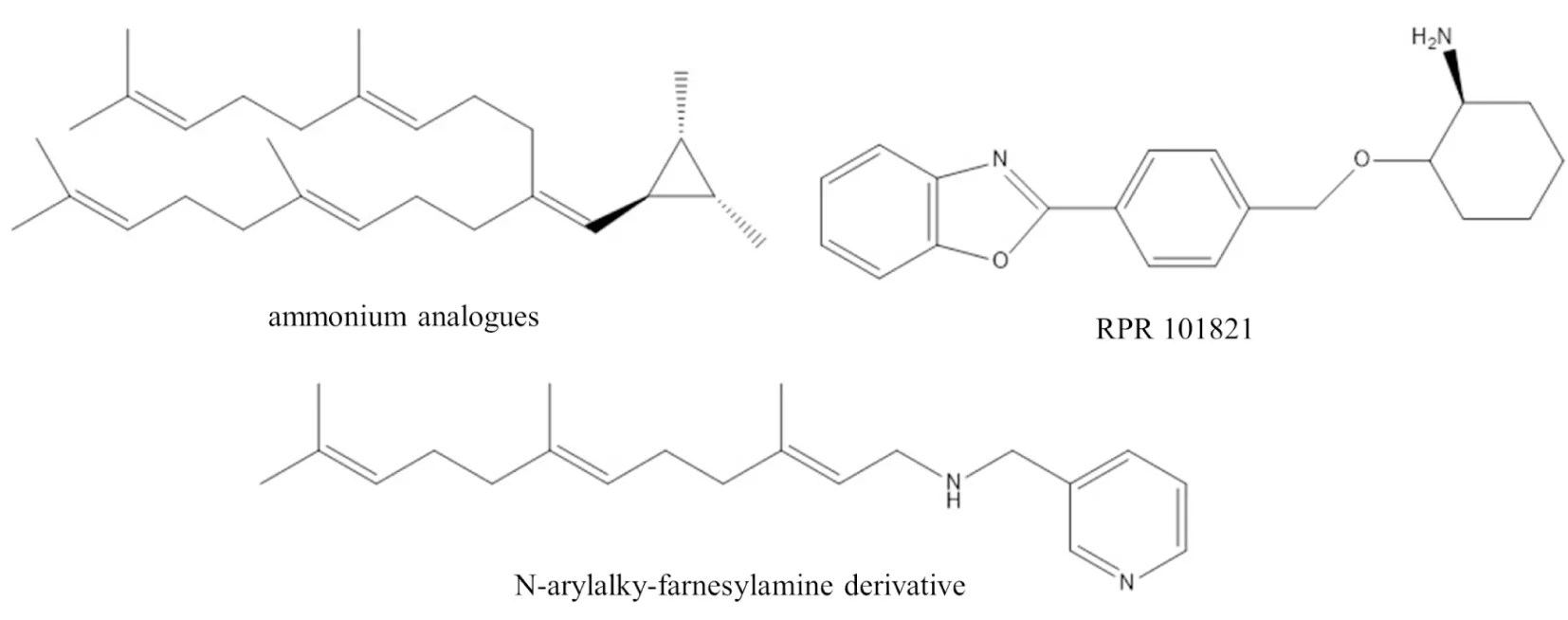
Fig 4 The chemical structures of ammonium analogues, RPR 101821 and N-arylalky-farnesylamine derivative
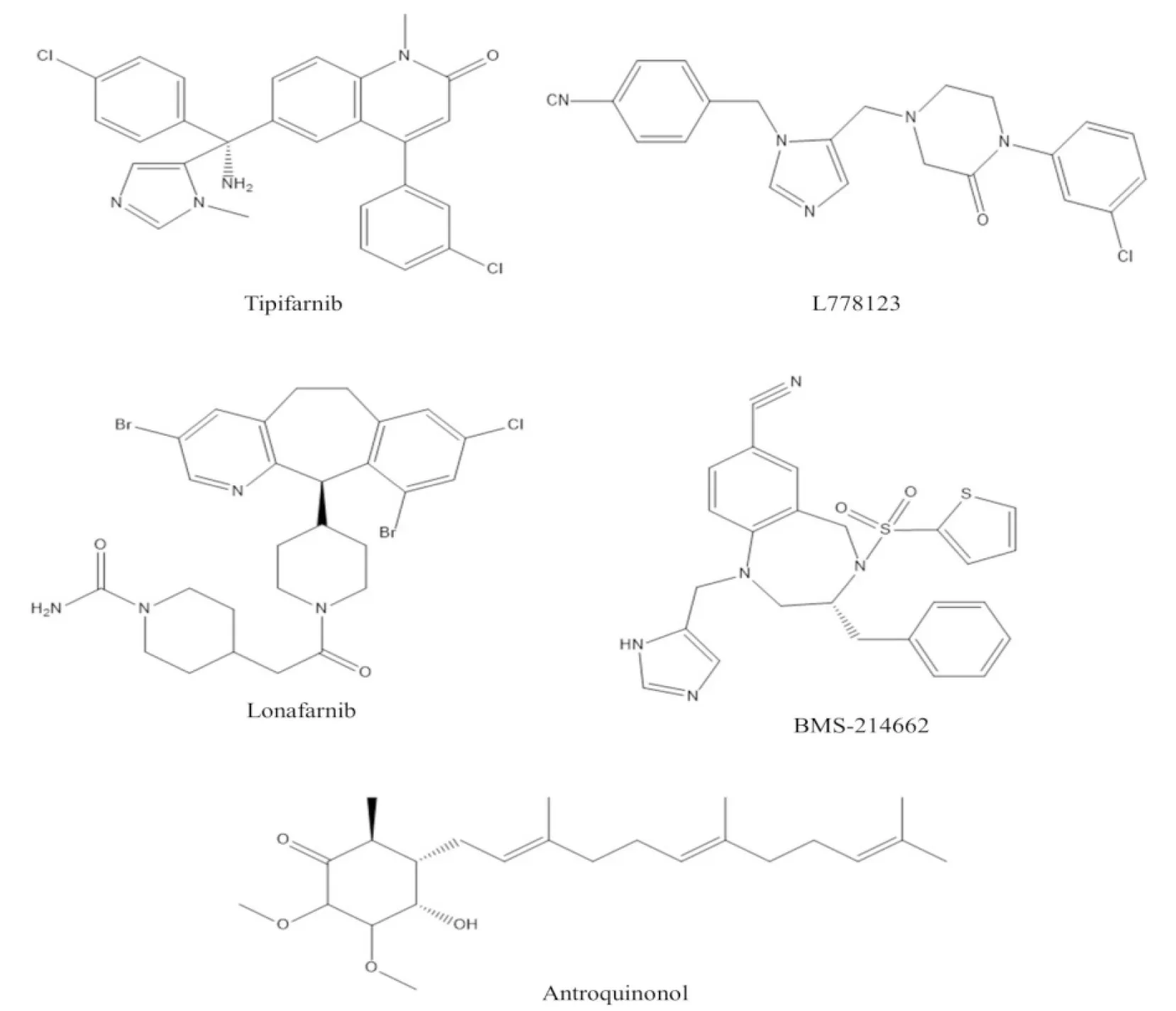
Fig 5 The chemical structures of tipifarnib, L778123, lonafarnib, BMS-214662 and antroquinonol
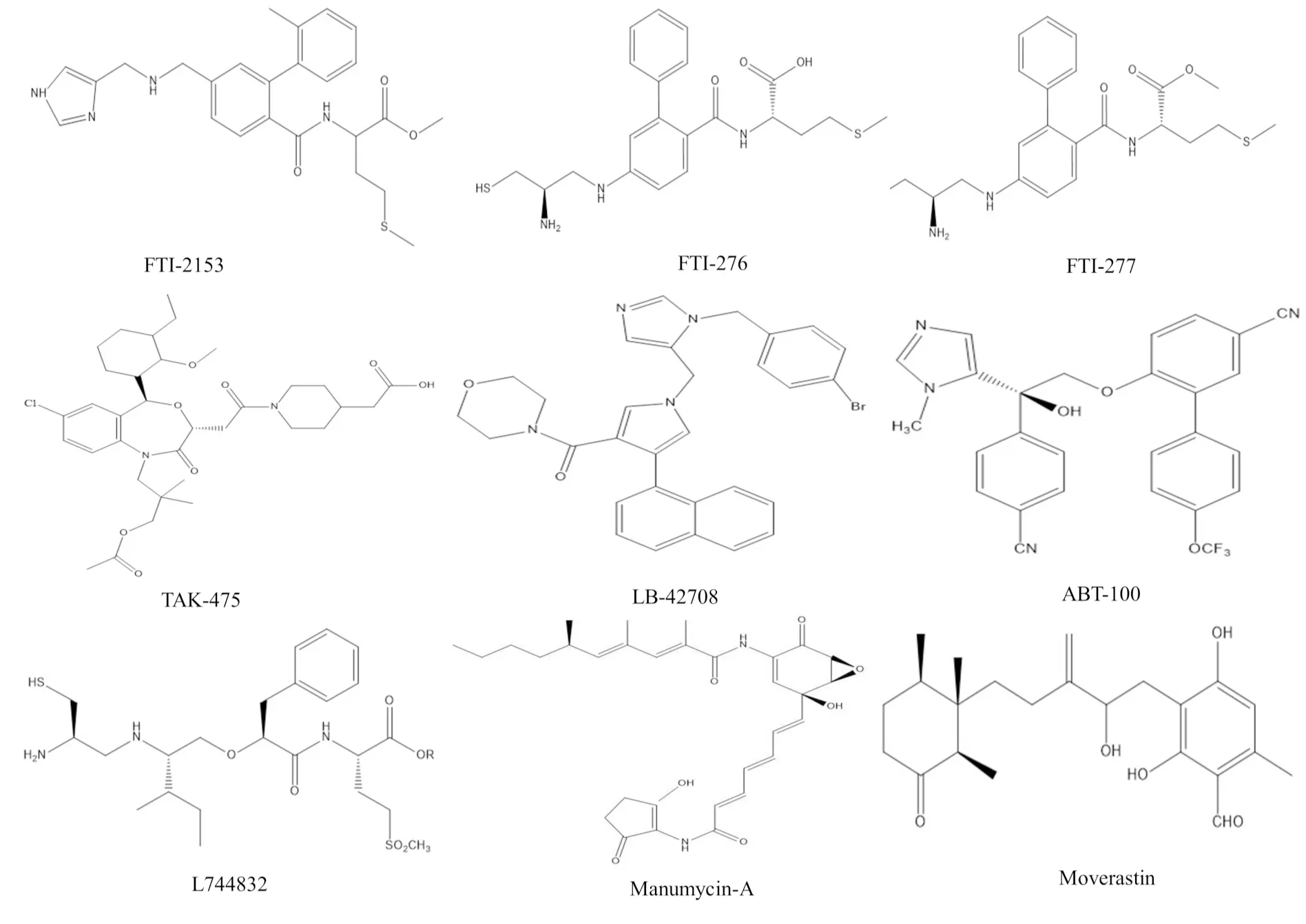
Fig 6 The chemical structures of FTI-2153, FTI-276, FTI-277, TAK-475, LB-42708, ABT-100, L744832, Manumycin-A and Moverastin
目前已有100多種各類FTIs被發現[8,18],其中5種化合物替吡法尼、洛那法尼、安卓奎諾爾、BMS-214662和L778123已進入臨床實驗研究階段或已完成臨床實驗。近期,替吡法尼和洛那法尼已被美國食品藥品監督管理局(Food and Drug Administration,FDA)批準分別用于T細胞淋巴瘤和早衰癥的治療。
3.1 FTase的底物模擬物底物蛋白羧基端的CAAX四肽基序及法尼基焦磷酸(FPP,法尼基的活化形式)是FTase最直接的底物。在FTase的催化作用下,其可將法尼基共價連接到CAAX四肽基序中半胱氨酸巰基上。基于這一機制,當前已合成或發現諸多底物模擬物,這些模擬物可以與CAAX四肽基序或法尼基焦磷酸競爭結合FTase的活性中心,從而抑制FTase對底物的催化作用。
除了上文所述的CVIM和CVFM外,其它FTIs活性較好的底物模擬物還包括isoprenyl phosphinylformates、ER-28488和ER-27856等,其中后3種化合物屬于FPP的類似物。ER-28488及其前體ER-27856對FTase的半抑制濃度(IC50)分別為3.6 nmol·L-1和39 μmol·L-1[6]。
3.2 過渡態模擬物在FTase催化法尼基連接至半胱氨酸巰基上時,半胱氨酸會形成一個碳正離子中間體,一些能夠很好模擬碳正離子中間體的化合物如銨類似物 (ammonium analogues)、N-芳烷基-法尼胺衍生物(N-arylalky-farnesylamine derivative)和RPR 101821(一個質子化的環已胺衍生物),它們能夠與碳正離子中間體競爭結合FTase的酶活性中心,進而抑制FTase活性[6,19]。
3.3 其它化合物除了底物模擬物和過渡態模擬物,還有許多不能歸屬于上述兩類模擬物的化合物也表現出較強的FTIs活性,這些化合物包括替吡法尼、洛那法尼、安卓奎諾爾、BMS-214662、L778123、L744832、TAK-475、Manumycin-A、FTI-276、FTI-277、FTI-2153、LB-42708、Moverastin和ABT-100等,其中前5個化合物已進入臨床實驗研究階段或已完成臨床實驗。值得注意的是,替吡法尼和洛那法尼已于2020年被FDA批準分別用于T細胞淋巴瘤和早衰癥的臨床治療。
3.3.1替吡法尼 替吡法尼是一種強效的FTase特異性抑制劑,其對FTase的IC50值可低達7.9 nmol·L-1。有研究檢測替吡法尼對53種常見人類腫瘤細胞系增殖的抑制效果,發現75%的腫瘤細胞系均對替吡法尼敏感,而且絕大多數對替吡法尼敏感的腫瘤細胞系均發生了H-Ras或N-Ras突變[20]。臨床研究結果顯示,在已接受替吡法尼單藥療法的重度預防治療的T細胞淋巴瘤患者中,客觀緩解率(objective response rate,ORR)約為50%。而在攜帶殺傷細胞免疫球蛋白樣受體(killer cell immunoglobulin-like receptor,KIR)突變體的患者中,替吡法尼的抗腫瘤活性明顯增強,這些患者的ORR可達70%,完全緩解率(complete remission,CR) 達40%[21]。于2020年,替吡法尼已被FDA批準用于T細胞淋巴瘤的臨床治療。
此外,應用替吡法尼治療膀胱癌[22]、唾液腺癌[23]、慢性粒細胞白血病、甲狀腺癌[24]、乳腺癌、膠質母細胞瘤和肺癌等腫瘤正處于各期臨床實驗階段。
3.3.2洛那法尼 洛那法尼也是一種特異性FTase抑制劑,有研究表明,其對7種常見腫瘤細胞的72 h平均IC50值約為8 μmol·L-1[25]。于2020年洛那法尼被FDA批準用于早衰癥的治療。因洛那法尼可以有效抑制FTase對早衰素 (presenilin) 的法尼基化,從而降低早衰素在細胞核中的積累[26]。此外,2018年FDA還授予洛那法尼用于治療丁型肝炎病毒 (HDV) 感染的突破性藥物資格(breakthrough therapy designation,BTD)[27]。
3.3.3安卓奎諾爾 安卓奎諾爾是一種萃取自臺灣特有珍稀物種牛樟芝(Antrodiacamphorata)的天然小分子化合物,它也是一種強效的FTase抑制劑[28]。有許多研究從不同角度探討了安卓奎諾爾抑制肺癌、乳腺癌、肝癌、結直腸癌和胰腺癌生長的分子機制[28]。機制研究發現,安卓奎諾爾的環狀結構可以直接結合于FTase活性中心的Ras-CAAX四肽基序結合位點,從而競爭性抑制Ras蛋白的法尼基化及其功能[28]。安卓奎諾爾對非小細胞肺癌(non-small cell lung cancer,NSCLC)的Ⅱ期臨床研究結果顯示,單獨使用安卓奎諾爾可提高肺癌的控制率,延長無進展生存期(progress free survival,PFS) 和總生存期(overall survival,OS)。雖然安卓奎諾爾對K-Ras突變陰性肺癌患者的治療效果明顯優于對K-Ras突變陽性肺癌患者的治療效果,但目前安卓奎諾爾仍然是治療K-Ras突變陽性肺癌患者唯一有效藥物[29]。此外,評價安卓奎諾爾的抗乙肝病毒(HBV)活性,探討安卓奎諾爾在慢性乙肝中的治療作用機制的Ⅱ期臨床實驗也正在進行中(Clinical Trials編號: NCT04112147和NCT036 25102)。其它曾經進入Ⅰ期臨床抗腫瘤研究的FTIs還包括BMS-214662和L778123,但由于存在較大的毒副作用,它們未能進一步進入更大規模的臨床研究。
3.3.4TAK-475等 TAK-475又名拉帕司他 (lapaquistat),一度進入Ⅱ期和Ⅲ期臨床研究,但由于其較強的肝臟毒性,在臨床實驗過程中被FDA叫停。此外,化合物L744834、Manumycin-A、FTI-276、FTI-277、FTI-2153、LB-42708、Moverastin和ABT-100等也具有較強的FTIs活性,目前它們正處于臨床前抗腫瘤活性評估和抗腫瘤作用機制研究階段[30]。
4 總結與展望
諸多研究表明,Ras蛋白在眾多腫瘤的病理進程中發揮著重要作用,而FTase通過法尼基化將Ras蛋白固定于細胞膜是Ras蛋白活化所必需的。因此,FTase可在Ras介導下促進腫瘤的發生發展。因Ras蛋白結構的特殊性,致使其缺乏合適的小分子作用位點,所以很長時間以來Ras蛋白一直被認為是“無藥可靶向的”。為了能夠找到用于治療Ras突變所誘發腫瘤的藥物,FTase及其抑制劑逐漸成了研究的焦點。在過去20多年,共有100多種各型FTIs被發現,其中5種FTIs包括替吡法尼、洛那法尼、安卓奎諾爾、BMS-214662和L778123已進入或已完成臨床實驗,替吡法尼和洛那法尼已于2020年被FDA批準分別用于T細胞淋巴瘤和早衰癥的臨床治療。
然而,還有很多FTIs止步于Ⅱ期臨床研究或未進入臨床研究,主要是因為這些FTIs表現出較強的毒副作用,包括嚴重的對正常細胞的毒性、肝臟毒性、胃腸不適和神經毒性等。基于對這些FTIs結構及其抑制FTase作用機制的認識,當前許多工作正在開展對這些FTIs進行結構改造或聯合用藥等方面的研究,以期提高這些FTIs的藥效及降低其毒副作用。同時,隨著新的化合物被合成、新的天然化合物被不斷發現及高通量篩選技術的日趨完善,許多新的FTIs先導或候選化合物將會被識別鑒定,這將會為Ras突變所誘發腫瘤等疾病的治療提供更多選擇。
