高效液相色譜法測定血漿中甘草西定濃度
2021-07-31 02:40:18王牧原
錦州醫科大學學報 2021年3期
王牧原
(錦州醫科大學附屬第三醫院,遼寧 錦州 121000)
甘草在我國是一種古老的藥用植物,甘草的根莖具有顯著的抗病毒、抗炎、抗癌、抗過敏、保肝和雌激素活性[1-3]。甘草西定是甘草的主要成分之一,其具有有很強的抗菌[4-5]、抗癌[6]、抗炎[7]活性。此外,甘草西定可防止UVA誘導的人皮膚成纖維細胞的光老化[8]。在研究甘草西定藥代動力學特征之前,需要測定甘草西定的血藥濃度,然而甘草西定的血漿濃度測定至今沒有相關報道。高效液相色譜法(high performance liquid chromatography,HPLC)是一種重要的檢測方法[9-11],其能夠定量檢測藥物在血漿中的濃度。本研究利用HPLC對甘草西定血漿濃度進行測定,并對方法學進行了驗證,以期為今后甘草西定藥代動力學研究提供分析方法,見圖1。
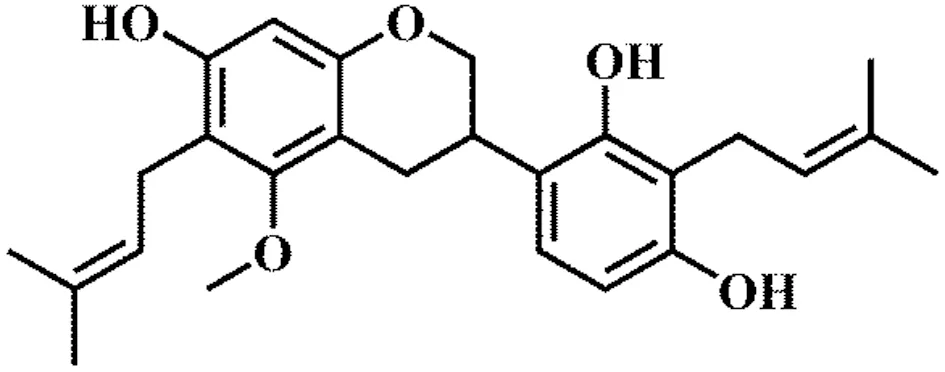
圖1 甘草西定結構式
1 儀器與材料
1.1 儀器
日本島津 LC-20AB高效液相色譜儀,其中包括CTO-20AB柱溫箱,SIL-20A自動進樣器和 SPD-20A紫外檢測器;UV752紫外可見分光光度計(上海諾科);貝克曼高速離心機(美國Beckman Coulter公司);XB220A萬分之一電子天平(瑞士Precisa公司);HERMO.SHAKER金屬混勻儀(英國Grant儀器有限公司);Synergy Uv純水儀(美國Millipore公司),氮吹儀(CM-12,北京成萌偉業科技有限公司),可調移液器(eppendorf,德國)等。
1.2 試劑
甘草西定對照品由云南西力生物技術股份有限公司提供,純度98%;乙腈和甲酸都是色譜級(德國默克);其他試劑為分析純,水為純化水。
2 方法與結果
2.1 標準溶液的配制
標準貯備液的配制:精密稱取甘草西定對照品2 mg,置10 mL量瓶中,用乙腈定容至刻度,得濃度為0.2 mg/mL的貯備液,置4 ℃冰箱保存,臨用時稀釋至所需濃度。
標準工作液的配制:將甘草西定標準貯備液用乙腈稀釋定容,配制成質量濃度分別為100、50、10、5、1、0.5、0.1、0.05、0.01 μg/mL 的甘草西定標準工作液。
2.2 測定波長的選擇
配制10 μg/mL甘草西定溶液2 mL,用乙腈作為對照液,在200~400 nm波長范圍內掃描,甘草西定在207 nm 和 285 nm處有等吸收,為了降低溶劑效應我們選擇285 nm作為甘草西定的測定波長。
2.3 色譜條件
采用Hypersil BDS C18反相分析色譜柱(大連依利特分析儀器有限公司,5 μm,4.6 mm×150 mm),柱溫設置40 ℃,流動相由0.1%的甲酸水和乙腈組成梯度洗脫。流速為 1 mL/min,檢測波長為285 nm,進樣體積10 μL,見表1。
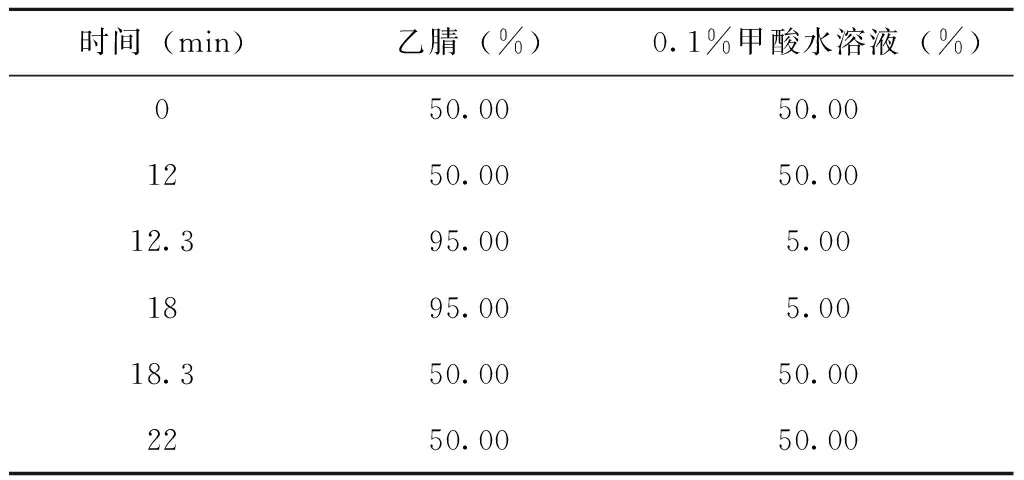
表1 甘草西定分析的梯度洗脫程序
2.4 血液樣品處理
取血漿0.3 mL,加10 μL甘草西定貯備液(0.2 mg/mL),混勻后加乙酸乙酯1 mL,振搖3 min后離心(10 000 r/min,15 min),取出有機相,用氮氣流吹干,100 μL 乙腈溶解殘渣,10 μL進樣分析。
2.5 方法的專屬性試驗
試驗分為3組,50 ng/μL標準液直接進樣組、空白血漿組和含甘草西定血漿組(50 ng/μL)。空白血漿組和含甘草西定血漿組按“2.3”方法進行血漿樣品處理。3組在“2.2”項色譜條件下,分別直接進樣。甘草西定保留時間為15.34 min,雜質沒有干擾,見圖2。
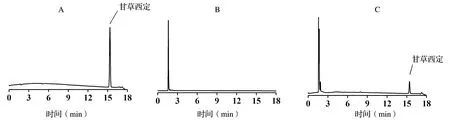
A:甘草西定對照品溶液;B:空白血漿;C:含甘草西定血漿
2.6 標準曲線的制備
取空白血漿0.5 mL,精密加入一定量的甘草西定標準溶液,使血漿中甘草西定的濃度分別為 0.1、0.3、0.5、1、3、10 μg/mL。并按“2.4”項下方法操作,記錄色譜。分別以甘草西定的濃度(X)為橫坐標,峰面積(Y)為縱坐標作直線回歸,得直線回歸方程分別為:Y=26865X-8199,R2=0.9993。結果表明,甘草西定檢測濃度在0.1~10 μg/mL范圍內與其峰面積線性關系良好。按信噪比S/N=1/2計算,最低檢測限為0.05 μg/mL。
2.7 精密度和回收率
配制濃度為0.1、1、10 μg/mL的甘草西定標準血漿樣品各5份,按“2.4”項下處理,于同一天內測定血樣中甘草西定的濃度,求得該方法的日內精密度,另將測定值除以理論值即為本測定方法的相對回收率,血漿測定的峰面積除以相同濃度化學樣品直接進樣的峰面積比值即為樣品的絕對回收率。同法配制甘草西定高、中、低的樣品各5份,置冰箱冷凍,自配制之日起,每日取出1份測定甘草西定的濃度,計算每種濃度樣品的 RSD 值,求得日間精密度。精密度及回收率試驗結果,日內RSD < 10%,日間精密度<15%,見表2。
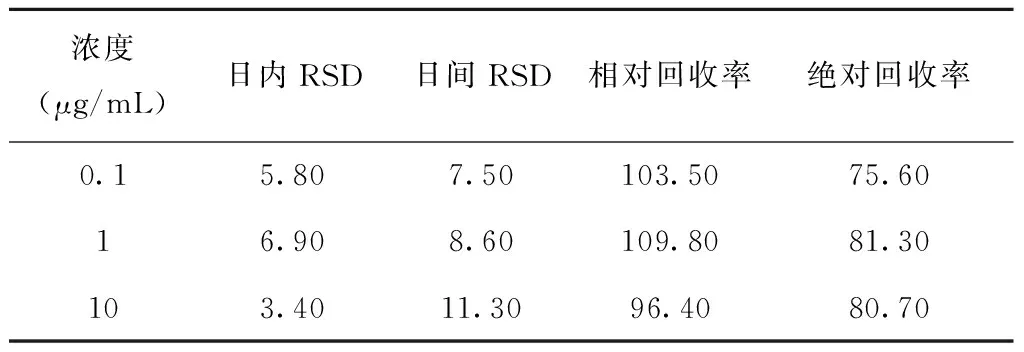
表2 精密度及回收率試驗結果(%)
2.8 乙腈中甘草西定在室溫的穩定性試驗
將0.1、1、10 μg/mL甘草西定標準品溶液在室溫下放置第0、8、16、24小時后,按“2.3”色譜條件用高效液相色譜測定其濃度,進樣量為10 μL。顯示甘草西定乙腈溶液在室溫下24小時內穩定性較好,見表3。
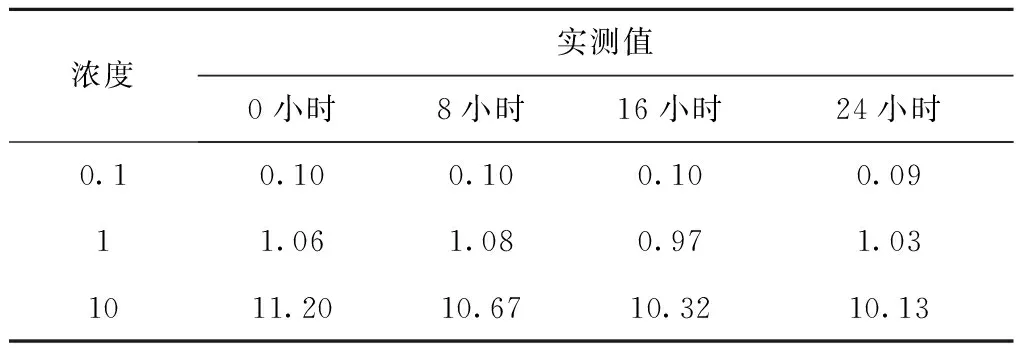
表3 不同時間乙腈中甘草西定的濃度(μg/mL)
2.8 甘草西定工作液在低溫條件長期保存的穩定性試驗
配制0.1、1、10 μg/mL甘草西定工作液,放入-20 ℃冰箱中冷凍,分別存放0、1、2和4個月后取出測其濃度,考察甘草西定在長期保存過程中的穩定性。結果顯示長期保存并不影響甘草西定的穩定性,見表4。
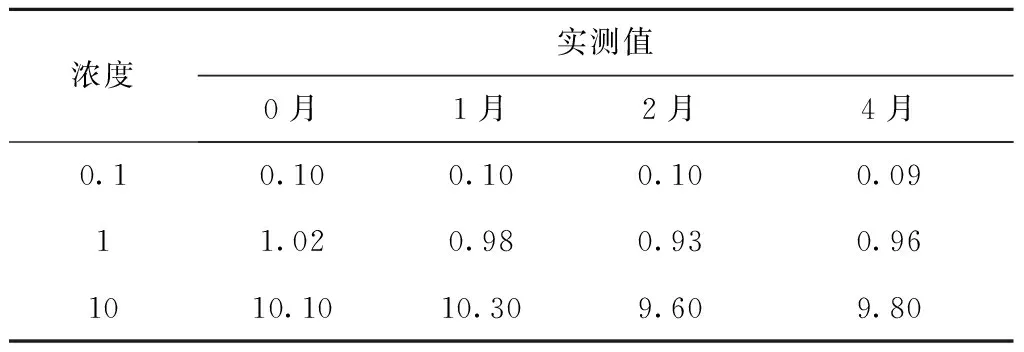
表4 甘草西定在乙腈中冰凍穩定性(μg/mL)
2.9 血漿中甘草西定多次凍融穩定性試驗
在空白血漿中加入適當甘草西定工作液,配制成0.1、1、10 μg/mL的甘草西定血漿樣品20 mL,然后各取0.5 mL分裝于離心管中,放入-20 ℃冰箱中冷凍,取出室溫解凍,連續凍融3次,每次按照血漿樣品處理后測含量,考察樣品凍融穩定性。結果顯示反復凍融3次并不改變血藥濃度,見表5。
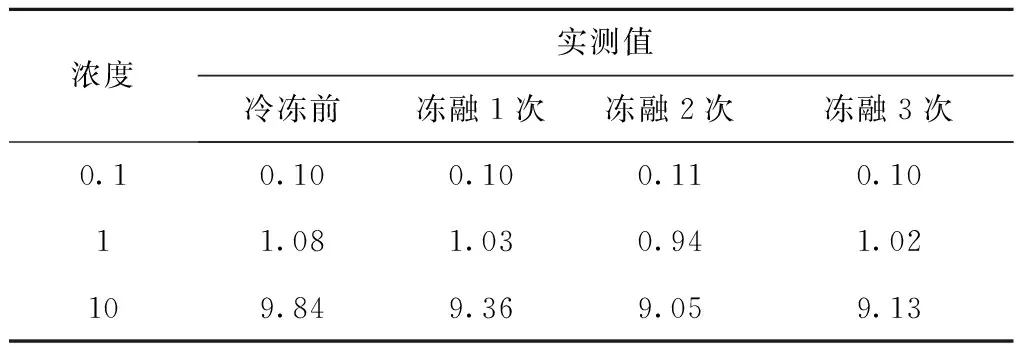
表5 凍融試驗結果(μg/mL)
2.10 甘草西定血漿中長期保存穩定性試驗
在空白血漿中加入適當甘草西定工作液,配制成0.1、1、10 μg/mL的甘草西定血漿樣品20 mL,然后各取0.5 mL分裝于離心管中,放入-20 ℃冰箱中冷凍,分別在第0、1、2和第4周取出,每次按照血漿樣品處理后測含量,考察樣品在長期保存過程中的穩定性。結果顯示長期保存并不影響血藥濃度,見表6。
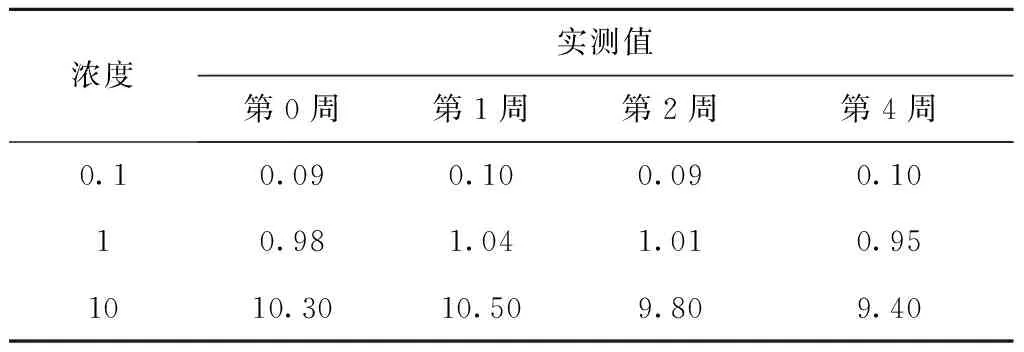
表6 甘草西定在血漿中的冰凍穩定性(μg/mL)
3 討 論
血藥濃度的檢測在藥物的研發和藥物的應用都具有重要的意義,如在藥物的研發階段通過血藥濃度來獲知藥代動力學參數從而為藥物的使用提供依據,在藥物的應用階段通過測定藥物在血漿中的濃度使藥物在達到治療效果同時避免不良反應的發生。常見的血藥濃度檢測的方法包括分光光度法、氣相色譜法、高效液相色譜法和免疫學方法。而高效液相方法是分析速度最快,應用范圍最廣的血藥濃度檢測方法[12-13]。
有關甘草西定血藥濃度的測定方法目前沒有相關文獻報道,本實驗采用HPLC 紫外檢測方法,首先用紫外分光光度計掃描甘草西定標準品,測得最大吸收波長為285 nm。因為甘草西定在乙腈中溶解性較好,因此流動相選用乙腈和0.1%甲酸水。在流動相中加入甲酸是因為甘草西定結構中有3個酚羥基,加入甲酸能使甘草西定在流動相和色譜柱中以分子形式存在,峰型更好。另外,我們曾采用梯度洗脫,流動相是由乙腈和0.1%甲酸水組成,兩者的極性不同,通過改變兩者在流動相中的比例從而改變流動相的極性,各組分在色譜柱中都有較好的容量因子k,并使甘草西定和其他組分在較短時間內達到較好的分離效果,甘草西定的保留時間合適。
由于生物樣品的組成復雜,基質對待測物的干擾較嚴重,并且待測物在樣品中的含量較低。因此,如何能從復雜的生物樣品中快速、準確地提取和分離目標成分,并達到分析儀器的檢測標準,需要對生物樣品進行預處理。生物樣品的處理方法包括沉淀蛋白和有機溶劑萃取等方法[14-15]。沉淀蛋白方法操作步驟簡單,向血清或血漿中加入一定量的有機溶劑、無機鹽或酸性物質等,樣品中的蛋白質遇到這些物質會發生變性反應,導致蛋白沉淀,沉淀的蛋白質經高速離心去除。蛋白沉淀法特別適用于強極性藥物或兩性類藥物,這些藥物難以用有機溶劑從血漿中提取。由于乙腈和甲醇對液相色譜分析和液相色譜-質譜的兼容性好,所以最常用的沉淀蛋白的有機溶劑為乙腈和甲醇[16]。我們先試用高氯酸和乙腈等沉淀蛋白,離心后取上清液直接進樣的方法,結果表明回收率和檢測限不能滿足要求且在甘草西定出峰時間有雜質干擾。因而采用液液萃取方法從血漿中提取樣品,通過氮吹富集后回收率和檢測限可滿足測定要求,樣品峰沒有干擾。
綜上所述,本文首次建立了血漿中甘草西定的高效液相測定方法,該方法精密度高,重現性好,回收率符合要求,方法科學,結果準確,易于推廣。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
云南醫藥(2019年3期)2019-07-25 07:25:14
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
海南醫學(2016年8期)2016-06-08 05:43:00
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
醫學研究雜志(2015年9期)2015-07-01 17:28:15
