Rac 3通過MAGEA6/AMPKα途徑抑制肺腺癌A549細胞的自噬
2021-07-31 02:40:18杜天宇肖旭陽
錦州醫科大學學報 2021年3期
關鍵詞:肺癌
杜天宇,肖旭陽
(錦州醫科大學附屬第一醫院,遼寧 錦州 121000)
肺癌目前是我國最常見的惡性腫瘤,在中國每年肺癌新發病例73.33萬人,死亡病例61.02萬人,居惡性腫瘤發病率及死亡率的首位[1]。隨著人們體檢意識增強,肺腺癌的檢出率、發病率逐漸提升,因此肺腺癌的發生、發展、耐藥等基礎研究一直是腫瘤研究中的熱點。
Rac 3是一種分子量為21 kd的小分子蛋白,屬于Rac家族成員,該家族被認為是細胞骨架的重要調節因子,在神經發育及腫瘤中起到重要作用[2]。Rac 3在肺癌中表達增高,沉默Rac 3后影響肺腺癌A549細胞的侵襲、遷移、上皮間質轉化,抑制增殖并促進凋亡[3-5]。自噬是一種高度保守的生物學過程,在腫瘤生存條件惡劣時可以提供必要的養分供細胞生存,而另一方面自噬可誘導凋亡,進而導致腫瘤細胞的死亡。Rac 3能在多種細胞系內引起自噬[6],但是探討Rac 3對肺癌自噬影響的研究較少,相關機制仍不明確。
CRISPR/cas9是一種新興的基因編輯技術,在小向導RNA(small guide RNA,sgRNA)的作用下可以切斷DNA雙鏈,并在基因組受損修復過程中發生移碼突變,從而達到敲除基因的效果[7]。本研究使用CRISPR/cas9技術,敲除肺腺癌A549細胞中的Rac 3基因并構建穩定表達的細胞系,并建立Rac 3過表達細胞株,探討Rac 3對肺腺癌自噬的影響及機制。
1 材料與方法
1.1 細胞培養和主要試劑
1.1.1 細胞培養:肺腺癌細胞系A549購自中科院細胞庫,用含10%胎牛血清的RPMI-1640,于37 ℃、5% CO2的恒溫恒濕培養箱內進行培養。
1.1.2 主要試劑:Addgene購買質粒pSpCas9(BB)-2A-Puro (PX459,Plasmid #48139),Rac 3抗體夠自ABCam,SQSTM-1/p62、AMPKα購自萬類生物,LC3、Phospho-AMPKα、ERK 1/2、Phospho-ERK 1/2購自CST,AKT、Phospho-AKT、MAGEA6購自ProteinTech。
1.2 puro-cas9-Rac 3-sgRNA質粒的構建
1.2.1 sgRNA的設計與合成:通過在線工具(https://zlab.bio/guide-design-resources)設計2條sgRNA,分別位于第2、3外顯子,序列分別為:sgRNA1#:5’-CGGTGGGGATGTACTCTCCG-3’和sgRNA2#:5’-CGGGTCAGGAGGACTACGAT-3’。sgRNA的正義鏈及反義鏈由Takara公司合成,收到的2對寡核苷酸鏈常規退火。
1.2.2 sgRNA載體構建:PX459質粒使用BbsI酶37 ℃酶切1 h,T4連接酶16 ℃過夜連接,導入到感受態細胞DH5α凃板,挑菌提取質粒,BbsI酶切后產物進行瓊脂糖凝膠電泳,選取不含酶切位點的質粒送堿基測序驗證。
1.3 Rac 3敲除及過表達A549細胞株的建立
1.3.1 敲除質粒轉染及單克隆提取:質粒使用Lipofectamine 3000試劑進行轉染,用篩選培養基(嘌呤霉素濃度1 μg/mL)篩選72 h后轉為維持濃度繼續培養,鏡下觀察單個細胞形成約含50個細胞的細胞集落時,消化稀釋細胞濃度至0.5個/微升,按2 微升/孔接種到96孔板內,標記出單個細胞的孔并加培養基繼續培養,約7 d后對標記的單克隆消化、擴大培養及鑒定。
1.3.2 Rac 3敲除細胞株的鑒定:根據sgRNA位置設計PCR引物,分別為:sgRNA1#(5’-CTTAGGTCGCCACGGATCTG-3’,5’-CTAGAACTCTGGCCAGCACC-3’)和sgRNA2#(5’-GGTGCTGGCCAGAGTTCTA-3’,5’-GGCTCACCAGAGAGAAGCAG-3’)。PCR產物使用T7EI酶于37 ℃反應20 min后進行瓊脂糖凝膠電泳,選取存在編輯的樣本送生物公司測序。
1.3.3 Rac 3過表達慢病毒感染A549細胞:Rac 3過表達慢病毒HBLV-h-RAC3-3xflag-NEO由上海漢恒公司包裝,A549細胞接種于96孔板,向孔內加入慢病毒,隨后用含G418培養基(800 μg/mL)進行篩選,72 h后降至維持濃度并擴大培養。
1.4 EBSS饑餓法誘導自噬
待檢測細胞按每個平皿按照1×106接種入10 cm培養皿中,至細胞融合約70%時棄液,PBS清洗2次后加入EBSS 10 mL,于37 ℃ 5%CO2的孵箱內,培育4 h后提取蛋白進行鑒定。
1.5 Western Blot檢測蛋白表達
待檢測的各細胞株在細胞融合至約70%時,用含PMSF及磷酸酶抑制劑的RIPA細胞裂解液提取蛋白,并采用BCA法進行定量。各組進行SDS-PAGE電泳后將蛋白轉移至PVDF膜上,5%BSA封閉,4 ℃搖床過夜孵育一抗,二抗室溫孵育,ECL化學發光法進行顯影。
1.6 統計學分析
采用GraphPad Prism 7.0軟件進行統計學分析,采用t檢驗進行比較,以P<0.05為差異有統計學意義。
2 結 果
2.1 sgRNA的設計
從NCBI獲得人源Rac 3蛋白的cDNA,使用在線工具分別于第2、3外顯子上設計sgRNA1#和sgRNA2#,確保當基因編輯導致移碼突變時,所有的Rac 3轉錄本都不能正確翻譯,以達到敲除的效果,見圖1。

圖1 通過在線工具設計sgRNA序列
2.2 Rac 3敲除的驗證
將含有puro-cas9-sgRNA1#和puro-cas9-sgRNA2#的質粒轉染入A549細胞中,單克隆篩選后進行T7E1酶切實驗。由sgRNA1#、sgRNA2#介導的Rac 3敲除細胞株分別記為KO1、KO2。T7E1酶切實驗中KO1和KO2細胞株的PCR產物都被切成了大小不同的兩條帶,證實基因組DNA存在錯配,即兩種sgRNA都對基因組DNA進行了編輯。將發生基因編輯的KO1和KO2細胞株送測序,提示KO1存在2個堿基丟失,KO2存在35個堿基丟失,均達到移碼突變的效果,見圖2。

圖2 T7E1酶切實驗及Sanger測序驗證Rac 3基因編輯
2.3 Western Blot驗證Rac 3的敲除及過表達
對KO1、KO2兩珠Rac 3敲除細胞株及慢病毒過表達Rac 3細胞株(OE)提取蛋白進行Western Blot檢測,提示與野生型A549細胞(WT)相比,KO1、KO2細胞株的中Rac 3蛋白被完全敲除了,OE細胞株表達Rac 3-3xflag,且Rac 3表達水平明顯高于WT組,見圖3。

**P<0.01
2.4 EBSS誘導自噬后LC3、SQSTM-1/p62的表達
經過EBSS誘導自噬4 h后提取蛋白。與未誘導的WT組A549細胞相比,EBSS誘導后各細胞株的LC3 II及LC3 II/I的比值升高。EBSS誘導后,KO1和KO2兩組的LC3 II、LC3 II/I比值均高于WT組,SQSTM-1/p62低于WT組,OE組的LC3 II、LC3 II/I比值低于WT型,差異均具有統計學意義。提示在肺腺癌A549細胞中,Rac 3敲除后自噬被激活,Rac 3過表達自噬受到抑制,見圖4。

*P<0.05;**P<0.01
2.5 雙向調控Rac 3后相關蛋白表達的變化
EBSS誘導自噬后,對各組進行Western Blot檢測活化的caspase3、MAGEA6、AKT及其磷酸化蛋白、ERK 1/2及其磷酸化蛋白、AMPK及磷酸化蛋白的變化。在Rac 3敲除后,MAGEA6表達下降,AMPKα及磷酸化AMPKα的表達增強,caspase3活化增強;在Rac 3過表達后,MAGEA6表達升高,AMPKα及磷酸化AMPKα表達降低,caspase3活化減弱。Rac 3表達出現變化后AKT和ERK1/2變化不顯著。提示EBSS誘導自噬,Rac 3敲除可以促進凋亡,并且降低MAGEA6的表達,促進AMPKα的表達及磷酸化水平,進而促進細胞自噬,而Rac 3過表達則相反,見圖5。
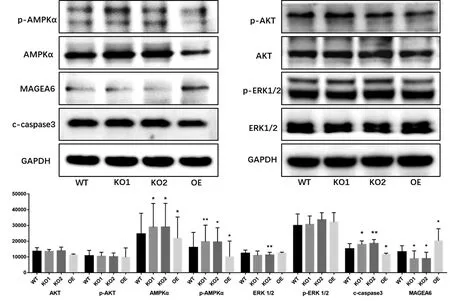
*P<0.05;**P<0.01
3 討 論
本研究通過CRISPR/cas9技術敲除肺腺癌細胞系A549中的Rac 3基因,使用慢病毒感染的方法過表達了A549細胞中的Rac 3基因,再以EBSS誘導自噬的方法,確定了Rac 3抑制A549細胞的自噬,并初步明確了Rac 3是通過MAGEA6/AMPKα途徑影響肺腺癌自噬。簇狀規則間隔的短回文重復序列(CRISPR)是一種廣泛存在于微生物內的適應性免疫系統,在進行工程化后的CRISPR/cas9技術具有快速、低成本、高效、拓展性高等特點[7]。CRISPR/cas9技術是通過設計sgRNA引導cas9蛋白靶向切割基因組DNA達到DNA雙鏈斷裂,隨后細胞以非同源末端連接的方式修復基因組DNA,在這一過程中有概率發生堿基的插入或丟失并導致移碼突變,從而達到敲除基因的效果[8]。sgRNA序列通常為在線工具所設計,因存在與其他基因相似序列結合的可能,故CRISPR/cas9基因編輯存在脫靶的風險并導致實驗結論的錯誤[9]。本研究通過設計兩個位于不同外顯子的sgRNA,并設立過表達組來減少該類誤差。
Rac 3是小分子GTPase,這類蛋白常在細胞中起到分子開關的作用,目前已知其在腫瘤及神經發育中起到重要作用。既往的研究表明,在膀胱癌中Rac 3通過PYCR1/JAK/STAT通路促進增殖、遷移和侵襲性[10],在肺癌中沉默Rac 3能抑制細胞增殖及誘導細胞凋亡[4]3061-3065,并且Rac 3可以通過p38 MAPK途徑調節細胞遷移、侵襲和EMT[3]2511-2522,在乳腺癌中沉默Rac 3導致侵襲性減少[11],Rac 3過表達誘導促生長和促遷移的基因,并且提示ERα陽性乳腺癌預后不良[12]。Rac 3在結腸癌中可以維持和誘導腫瘤細胞干性,并可以誘導非腫瘤細胞向腫瘤轉化[13]。Rac 3還可以增強宮頸癌細胞對順鉑的耐藥[14]。自噬是目前的一個研究熱點,在腫瘤細胞中自噬既可以保護腫瘤生存,同時也可以促進凋亡,而Rac 3被發現在多種細胞系中可以抑制自噬[6]35291-35298。我們研究發現在A549細胞中敲除Rac 3可以促進LC3 I型向II型轉化、p62積累水平降低及促進caspase3活化,而相反的過表達Rac 3抑制LC3 I型向II型轉化并抑制caspase3活化。這提示Rac 3對肺腺癌的自噬及凋亡起抑制的作用。在前期的預實驗中,我們對Rac 3敲除及過表達的細胞株做了RNA-Seq,篩選得到4個差異基因ADAMTS2、GABRA3、MAGEA6及CSRNP3,通過預實驗驗證及查詢文獻,我們推測Rac 3對自噬的抑制與MAGEA6基因有關。
MAGEA6是黑色素瘤抗原家族的成員,該家族成員在真核生物中保守,在腫瘤中常被異常激活。MAGEA6沉默在膠質瘤、腎細胞癌等多種腫瘤中直接上調AMPKα表達及磷酸化并抑制mTORC1通路[15-16]。MAGEA3/6可以特異性的結合TRIM28 E3泛素連接酶,導致AMPKα的泛素化降解[17]。在我們的研究中觀察到,當Rac 3敲除后MAGEA6表達隨之降低,這導致了AMPKα泛素化降解的減少,AMPKα及其磷酸化水平的增高。AMPKα磷酸化水平增高會抑制mTORC1的活性,從而解除了mTOR通路對自噬的抑制,導致自噬的增強[18-19]。當過表達Rac 3后,MAGEA6表達隨之升高,這導致AMPKα降解的增加及活性降低,從而導致mTOR通路被激活,抑制自噬。在肺癌等多種腫瘤中,Rac 3表達水平上升,這也就導致MAGEA6的高表達,進而抑制AMPKα并激活mTOR通路,導致自噬被抑制,從而達到減少凋亡、促進細胞成活的目的。但是Rac 3是如何調控MAGEA6的機制目前尚不確認,有待于進一步研究。
綜上所述,我們發現Rac 3可以上調MAGEA6的表達水平,抑制AMPKα表達及活性,激活mTOR信號通路,抑制自噬及凋亡,從而達到使肺腺癌細胞成活的目的。這提示Rac 3在肺腺癌中起到重要作用,可能會為肺癌的治療提供新的靶標。
猜你喜歡
保健醫苑(2023年2期)2023-03-15 09:03:04
中國臨床醫學影像雜志(2022年2期)2022-05-25 13:24:34
中國藥學藥品知識倉庫(2022年1期)2022-03-23 04:16:57
昆明醫科大學學報(2021年4期)2021-07-23 01:21:44
天津醫科大學學報(2021年2期)2021-03-29 05:30:38
天津醫科大學學報(2019年6期)2019-08-13 07:04:26
癌變·畸變·突變(2016年3期)2016-02-27 06:15:34
醫學研究雜志(2015年12期)2015-06-10 06:57:46
中國當代醫藥(2015年7期)2015-03-01 02:01:19
鄭州大學學報(醫學版)(2015年1期)2015-02-27 14:50:26
