共軛體系的分類及結構特征
2021-07-14 02:47:06朱秋華
大學化學 2021年6期
關鍵詞:示意圖
朱秋華
南方醫(yī)科大學藥學院,廣州 510515
在有機化學中,共軛體系是指相鄰原子或基團的電子軌道發(fā)生重疊或者說發(fā)生相互作用,電子發(fā)生離域運動、體系能量降低、鍵長平均化的結構單元。共軛體系的存在不僅對化合物的物理性質有很大影響,如對紫外吸收光譜、熒光光譜、核磁共振譜的影響,也對化合物的反應活性及反應發(fā)生位置有很大的影響。因此,共軛體系不僅是有機化學課程中一個非常重要的內容,也是理論化學、材料化學、分析化學、藥物化學等多個領域研究的一個重要內容。共軛體系對化合物性質的影響源于其獨特的電子云密度分布變化或者說電子效應–共軛效應。不同原子或基團組成的共軛體系具有不同類型和程度的共軛效應。共軛效應是有機化學課程的重點和難點內容,掌握共軛體系的分類及結構特征,對理解化合物的共軛效應具有重要作用。但一般的有機化學教材及文獻對此沒有系統(tǒng)的介紹。一般大學有機化學教材只是在討論1,3-丁二烯獨特的結構與性質時,簡單地把共軛體系定義為:鍵長平均化,共軛體系中的各原子共平面并有一個垂直于該平面的p軌道,以及各原子上的p軌道互相平行并重疊的電子流域結構體系。而把碳氫σ單鍵(σCH)與π鍵或p軌道重疊而形成的電子流域體系稱為超共軛體系。隨著共軛體系的深入研究,共軛體系的內涵已遠遠超出了一般大學有機化學教材對共軛體系的定義,如文獻報道的空間共軛[1,2]、含飽和碳原子的芳香體系[3]、碳-氟σ單鍵(σCF)參與的超共軛[4]等。本文綜述了共軛體系的類型及結構特征,以拓展共軛體系在有機化學教學中的內涵,加深師生對共軛體系及共軛體系對化合物結構與性質影響的理解。因為共軛體系與π鍵和σ鍵有關,本文在介紹共軛體系的分類及結構特征前,先簡單地介紹π鍵與σ鍵在價鍵(valence bond,VB)理論以及分子軌道(molecular orbital,MO)理論中的表示方法。然后通過實例的結構式、VB及MO理論的電子軌道示意圖,闡明共軛體系的分類及其結構特征。
1 VB及MO理論中的共價鍵形成示意圖
共軛體系一般是根據(jù)參與共軛的電子軌道類型來進行分類。為了更好地理解共軛體系的分類及結構特征,我們需要先了解一下VB及MO理論對共價鍵的定義及電子軌道示意圖。VB理論認為共價鍵的本質是原子相互接近時,原子價層的電子軌道(n電子軌道)重疊(波函數(shù)疊加),原子間通過共用自旋相反的電子對,使能量降低而形成的化學鍵。原子軌道重疊具有方向性,軌道以頭碰頭方式重疊形成電子云呈鍵軸對稱的σ鍵,以肩并肩方式重疊形成電子云呈面對稱的π鍵。圖1a為原子的n電子軌道(s、p和spx雜化電子軌道,x= 1–3)重疊形成σ鍵和π鍵的示意圖。圖中原子軌道重疊示意圖和成鍵軌道圖是σ鍵和π鍵的兩種表示方法,常常在討論反應機理、電子效應及共軛體系時使用。除了這兩種表示方法外,還有大家熟悉的短橫線“–”和電子對“˙˙”表示法。
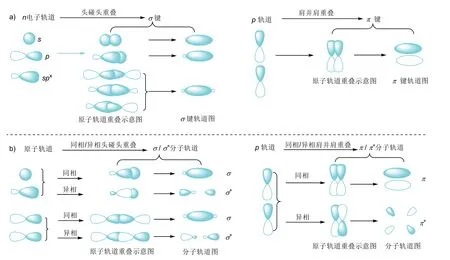
圖1 VB理論的σ和π鍵(a)以及MO理論的成鍵/反鍵分子軌道(σ和π/σ*和π*)形成示意圖(b)
MO理論認為原子形成分子后,電子不再屬于個別的原子軌道,而是屬于分子軌道。分子軌道是原子軌道的線性組合,有幾個原子軌道就可以組合成幾個分子軌道。類似VB理論中σ鍵和π鍵的形成,原子軌道以頭碰頭方式重疊形成電子云呈鍵軸對稱的σ分子軌道,而原子軌道以肩并肩方式重疊形成電子云呈面對稱的π分子軌道。形成分子軌道的原子軌道能量相近。波函數(shù)(原子軌道)同相重疊形成的分子軌道又稱為成鍵分子軌道,用σ和π表示,而原子軌道異相重疊形成的分子軌道稱為反鍵分子軌道,用σ*和π*表示。成鍵分子軌道能量比原子軌道能量低,反鍵分子軌道能量比原子軌道能量高。當電子進入成鍵分子軌道的數(shù)目比進入反鍵分子軌道的數(shù)目多時,能量降低,形成穩(wěn)定的化學鍵。圖1b為一個s軌道和一個雜化軌道同相/異相頭碰頭重疊形成σ/σ*分子軌道,兩個p軌道同相/異相頭碰頭重疊形成σ/σ*分子軌道,以及兩個p軌道同相與異相肩并肩重疊形成π/π*分子軌道的示意圖。對比MO理論中的σ和π成鍵分子軌道與VB理論中的σ和π鍵,可以看到二者的軌道圖及符號其實是相同的,只是名稱和含義有所不同。
2 共軛體系的分類
報道的共軛體系分類有三種,即基于電子軌道類型、共軛體系中相鄰原子或基團之間的連接方式以及電子軌道共軛程度大小的分類。
2.1 基于電子軌道類型的分類
相鄰原子或基團的電子軌道發(fā)生重疊是共軛體系最根本的結構特征。因此,共軛體系的分類一般是根據(jù)電子軌道的類型進行分類。從VB理論來看,共軛體系是由相鄰原子或基團的成鍵電子軌道(π鍵或σ鍵電子軌道)與成鍵或n電子軌道重疊所形成的能量降低結構體系;而從MO理論來看,共軛體系是由含一對電子的成鍵分子軌道(π或σ成鍵分子軌道)或n軌道與空的反鍵分子軌道(π*或σ*)或含0–1個電子的p軌道相互作用,形成能量更低的成鍵分子軌道和能量更高的反鍵分子軌道,進入能量更低的成鍵分子軌道電子數(shù)比進入能量更高的反鍵軌道電子數(shù)多,從而使能量降低的結構體系(共軛體系MO能級示意圖如圖2所示)。如表1所示,根據(jù)VB理論中的電子軌道類型,共軛體系可分為π–π、n–π、σ–π、σ–n和σ–σ五種類型,而根據(jù)MO理論中的電子軌道類型,可將共軛體系可分為π–π*、n–π*、σ–π*、π–p、n–p、σ–p、π–σ*、n–σ*和σ–σ*九種類型。
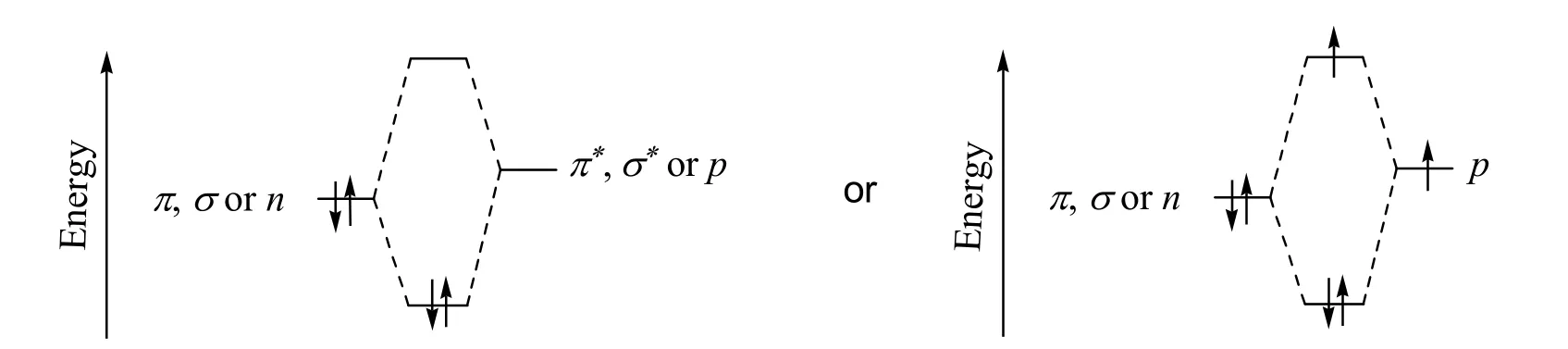
圖2 形成共軛體系的MO能級示意圖

表1 基于VB和MO理論中電子軌道類型的共軛體系分類
廣義的共軛體系是指由各種電子軌道重疊所形成的電子離域的結構體系,但一般把無σ鍵軌道參與的共軛體系稱為共軛體系,而把有σ鍵軌道參與的共軛體系稱為超共軛體系,如Alabugin等[5]在超共軛綜述中所述。根據(jù)VB理論中的軌道類型,共軛體系可分為π–π和n–π共軛,超共軛體系可分為σ–π、σ–n和σ–σ超共軛。而根據(jù)MO理論中的軌道類型,共軛體系分為π–π*、n–π*、π–p和n–p共軛,超共軛體系分為σ–π*、σ–p、σ–σ*、π–σ*和n–σ*超共軛。
2.2 基于相鄰共軛基團連接方式的分類
共軛體系中相鄰的原子或原子團可以是連在單鍵或雙鍵兩端的原子或基團、同一碳原子上的原子或基團、或者非價鍵直接相連但在空間上相鄰的原子或基團。一般把非價鍵相連但在空間上相鄰的原子或基團形成的共軛體系稱為空間共軛(through-space conjugation),而把由價鍵相連的鄰位或同位原子或基團形成的共軛體系稱為價鍵共軛(through-bond conjugation)。
2.3 基于共軛效應強弱的分類
Mulliken等[6,7]認為飽和與不飽和基團參與的共軛,只有量的區(qū)別而沒有性質的差別。建議將π與π鍵軌道的作用稱為一級共軛,σ和π鍵的相互作用稱之為二級共軛或者一級超共軛,而把σ鍵之間的作用稱之為三級共軛或二級超共軛。
3 共軛體系的結構特征
由于在有機化學教材中只提到了基于電子軌道類型的共軛體系分類,而未涉及基于共軛體系中相鄰原子或基團之間的連接方式以及電子軌道共軛程度大小的分類。并且,電子軌道的重疊是形成共軛體系的本質特性。因此,在以實例闡明不同共軛體系的結構特征時,以基于電子軌道類型分類的共軛體系,狹義共軛體系與超共軛體系,進行討論。
3.1 狹義共軛體系的結構特征
狹義共軛體系,即無σ鍵參與的共軛體系,包含通過價鍵形成的π–π和n–π共軛體系,以及通過空間(through-space,TS)形成的π–π和n–π共軛體系(TS-π–π和TS-n–π共軛體系)。由于通過價鍵形成的共軛體系比較普遍,而通過空間形成的共軛體系只在特殊結構的分子中存在,在討論體系類型時,除非指明是空間共軛,一般是指價鍵共軛。下面通過實例、簡單明了的VB理論軌道示意圖(見圖1a,π鍵用原子軌道重疊示意圖表示)闡明共軛體系的結構特征。
3.1.1π–π共軛體系結構特征
π–π共軛體系就是如圖3所示的重鍵(雙鍵或叁鍵)與單鍵間隔相連的分子結構單元。圖3a是比較大的π–π共軛體系,圖3b是最簡單的π–π共軛體系。在這些最簡單的共軛體系中,單鍵(標示紅色的鍵)在中間像一根扁擔一樣兩端各挑一個重鍵。1,3-丁二烯就是這樣一個最簡單的π–π共軛體系。如圖3c和3d所示,當我們把1,3-丁二烯分子中的兩個π鍵分別用兩個肩并肩的p軌道表示時,就會發(fā)現(xiàn),四個p軌道全部相鄰、平行。如圖3c所示,這四個p軌道是相互重疊的,其中1、2號碳上的兩個p軌道以及3、4號碳上的兩個p軌道的重疊程度大于2、3號碳上的兩個p軌道的重疊程度。但我們經(jīng)常用如圖3d所示的,用相互平行但不重疊的p軌道來表示肩并肩重疊形成了π鍵的兩個p軌道。實際上,只要相鄰碳原子上有平行的p軌道,它們就會重疊并形成如電子云呈面對稱的π鍵(見圖1中π鍵電子云圖)。也就是說,1,3-丁二烯分子形成了一個4原子4電子的大Π鍵(Π44)(Πnm,n為參與共軛的電子軌道數(shù),m為參與共軛的電子數(shù)[8])。四個碳原子上的p軌道里各含一個電子,這4個p軌道里的4個電子在這個大Π鍵中做流域運動,而不是局限在1、2位和3、4位碳原子之間,即1、2位和3、4位碳原子之間的電子云密度降低,使1、2位和3、4位之間的C=C雙鍵鍵長變長(0.137 nm,普通C=C雙鍵鍵長0.133 nm),而2和3位碳原子之間電子云密度增加,使2和3位之間的C-C單鍵鍵長變短(0.146 nm,普通C-C單鍵鍵長0.154 nm)。也就是說,是這種大Π鍵的存在使1,3-丁二烯的鍵長平均化。我們可以想到,這個π–π共軛體系有多長,就可以形成多長的大Π鍵,即共軛體系里的大Π鍵電子就可以在多長的范圍做離域運動。如圖3a所示的苯環(huán)分子是一個環(huán)狀的Π66共軛體系,其鍵長完全平均化(鍵長0.140 nm)。
另外,圖3e從MO理論中的成鍵分子軌道與反鍵分子軌道的相互作用以及MO能級示意圖解釋了1,3-丁二烯分子中的π–π共軛作用。如圖3e所示,分子中一個C=C之間的π成鍵分子軌道可以作為電子供體,而另一個C=C之間的π*反鍵分子軌道作為電子受體,π和π*分子軌道相互作用形成能量更低、運動范圍更大的成鍵分子軌道,電子進入該分子軌道,從而使體系能量降低,鍵長平均化。值得一提的是,這種MO軌道對于π–π共軛的解釋是把π–π共軛看成兩個π鍵的相互作用[5]。如果把分子中四個原子的p軌道重疊當成一個大的Π鍵用MO理論來討論,分子軌道示意圖、MO能級圖以及對π–π共軛體系能量降低,鍵長平均化的解釋有所不同[9]。

圖3 存在π-π共軛體系的幾個分子結構簡式(a和b)、以肩并肩平行的p軌道表示1,3-丁二烯(CH2=CH-CH=CH2)中π鍵的結構式(c和d)以及以π和π*分子軌道和MO能級示意圖表示的π–π*共軛作用(e)
3.1.2n-π共軛體系結構特征
n–π共軛體系是相鄰原子或基團的非鍵n電子軌道與π鍵電子軌道相互作用而形成的共軛體系。因為n電子軌道包含p軌道和spx雜化軌道,所以n–π共軛體系又可分為p–π和spx–π共軛。
3.1.2.1p–π共軛體系結構特征
p–π共軛體系是相鄰原子或基團的p軌道與π鍵軌道相互作用形成的共軛體系。按照參與共軛的p軌道里含電子的多少,又可以把p–π共軛分成:含2個電子、1個電子和0個電子的p軌道與π鍵形成的p–π共軛體系。由于一個原子軌道只能容納2個電子,含2個電子的p軌道可以稱為滿電子的軌道,而含1個電子和0個電子的p軌道可以分別稱為單電子和無電子的空軌道,所以用p(滿)–π、p(單)–π或者p(空)–π來分別表示含2個電子、1個電子和0個電子的p軌道與π鍵形成的p–π共軛體系。
如圖4所示的溴乙烯分子/烯丙基負離子、烯丙基自由基、丙烯基正離子分別存在p(滿)–π、p(單)–π或者p(空)–π共軛。Br原子價層有七個電子,其中一個電子與碳形成σ鍵后,還有三對未參與成鍵的電子,這些未成鍵的電子對也叫孤對電子,其中有兩對孤對電子位于p軌道中。碳負離子、碳自由基及碳正離子是反應過程中C-X鍵發(fā)生異裂或均裂形成的活潑中間體,異裂或均裂后的碳原子雜化軌道轉變成p軌道[9]。如圖4b所示,當我們把這些化合物中的π鍵用兩個肩并肩平行的p軌道來表示時,就可以很清楚地看到,溴原子/碳負離子、碳自由基及碳正離子上分別含一對電子、一個單電子和無電子的p軌道與π鍵中的p軌道相鄰并平行,即重疊形成了電子離域的3原子4電子(Π34)、3原子3電子(Π33)和3原子2個電子(Π32)的大Π鍵共軛體系。

圖4 存在p-π共軛體系的溴乙烯(BrCH=CH2)、烯丙基負離子(CH2=CHCH2-)、丙烯基自由基(CH2=CHCH2·)以及丙烯基正離子(CH2=CHCH2+)的結構式
3.1.2.2spx-π共軛體系結構特征
大家都知道,苯胺分子中N原子的堿性遠小于脂肪胺中N原子的堿性,即苯胺分子中N原子上孤對電子接收質子的能力,遠小于脂肪胺中N原子上孤對電子接收質子的能力。其原因是苯胺分子中N原子上的孤對電子與苯環(huán)中的π鍵形成了共軛。有機化學教材指出苯胺分子中N原子上的孤對電子是以介于sp2與sp3雜化軌道之間的雜化軌道參與共軛。可以肯定的是,無論是以什么樣的雜化軌道參與共軛,N原子上參與共軛的非鍵孤對電子軌道不可能與共軛的π鍵p軌道平行(如圖5a所示),即與有機化學教材中關于共軛體系中p軌道相互平行的定義是不相符的。顯而易見,雜化軌道與相鄰π鍵電子軌道的重疊程度要小于π–π、p–π共軛體系相鄰p軌道的重疊程度。
我們的實驗結果顯示,與苯相連的N原子可以以介于sp2與sp3雜化軌道之間的雜化軌道成鍵,也可以以sp2雜化軌道成鍵。如圖5b所示,在1,3-二苯基-4,5-二羧乙基四氫嘧啶(diethyl 1,2,3,6-tetrahydro-1,3-diphenylpyrimidine-4,5-dicarboxylate,TTHP)分子中,存在兩個與苯基相連的氮原子,即N1和N2。N1與三個碳原子形成的σ鍵鍵角均小于sp2雜化軌道鍵角120°,但大于NH3中N原子以sp3雜化軌道成鍵的鍵角106.8°,這說明N1原子如有機化學教材中提到的,是以介于sp2與sp3之間的雜化軌道成鍵。但該分子中的N2與三個碳原子形成的σ鍵鍵角之和剛好等于360°,且N2與相連的三個碳原子共平面,這說明N2原子是以sp2雜化軌道成鍵。值得一提的是,該N2原子sp2雜化軌道平面與苯環(huán)平面成一定的夾角,而與連有酯基的C=C雙鍵平面幾乎共平面。這說明N2的孤對電子主要與連有酯基的C=C雙鍵共軛。TTHP為我們發(fā)展的一個多組分反應產(chǎn)物,單晶結構發(fā)表在該合成方法文章中[10]。

圖5 苯胺(C6H5NH2)分子中的spx–π共軛(a)及N原子以sp2 (N2)和介于sp2與sp3之間的雜化軌道(N1)成鍵的實例(b)
3.1.3 空間共軛體系結構特征
空間共軛體系(TS-共軛)是指共軛的原子或基團之間沒有σ單鍵相連,但它們由于空間距離比較近,也像圖3–5中以σ單鍵相連的原子或基團一樣可以通過π與π或π與n軌道的重疊,形成電子流域的共軛體系。空間共軛不僅存在于分子內,也存在于分子間,即空間共軛可以分為分子內和分子間的TS-共軛體系。
3.1.3.1 分子內的TS-共軛體系結構特征
如圖6中的化合物1[11]和化合物2[2],其空間相鄰的苯基(紅色虛線相連的苯基)π鍵電子軌道重疊,形成了π–π空間共軛(TS-π–π)。而在化合物 THP-1 (diethyl 1,2,3,6-tetrahydro-1,2,3-triphenylpyrimidine-4,5-dicarboxylate)中,其空間相鄰的N原子上的孤對電子軌道與C=C中的π鍵電子軌道重疊,形成了n-π空間共軛體系(TS-n-π) (紅色虛線相連的N原子和C=C雙鍵)。

圖6 幾個分子內具有空間共軛的化合物
化合物的空間共軛可以從前線軌道電子云分布圖反映出來。化合物THP-1是我們在2011年發(fā)展的一個高效的合成一系列新型 C6位未取代四氫嘧啶類化合物(C6-unsubstituted tetrahydropyrimidines, THPs)的多組分反應產(chǎn)物[12,13]。如圖7所示,THP-1可以形成兩種分別發(fā)藍色熒光和綠色熒光的同質多晶(1b和1g)。雖然THP-1是共軛程度很低的非平面分子,但其熒光晶體THP-1b和THP-1g熒光量子率卻分別高到72%和93%[14]。發(fā)光機理研究發(fā)現(xiàn)THP-1的分子中存在空間共軛[14]。如圖7中最高占有分子軌道(HOMO)電子云分布圖HOMO-1b (II)和-(III)所示,N1上含孤對電子的雜化軌道與C=C雙鍵中的π鍵電子軌道在空間上相鄰并重疊(紅色圓圈標注的部分),形成了n–π空間共軛。N1原子上的三個共價鍵夾角分別為110.3°、117.5°和118.2° (conformation-1b-I),鍵角均小于120°,但大于106.8°,這說明以N1原子以介于sp2與sp3雜化軌道成鍵。

圖7 THP-1的分子結構以及兩種熒光異構體圖(1b和1g) (365 nm熒光燈下)、1b單晶中不同角度的構象(conformation-1b I–III)及1b單晶構象的HOMO電子云分布圖(HOMO-1b II–III)
3.1.3.2 分子間的TS-共軛體系結構特征
空間共軛不僅存在于分子內,還存在于分子間。香港科技大學唐本忠院士團隊仔細研究了幾個僅含游離苯基的小分子固態(tài)熒光產(chǎn)生機理,如研究了1,1,2,2-四苯基乙烷分子在溶液中的熒光發(fā)射光譜波長僅為290 nm,而固態(tài)的熒光發(fā)射光譜波長為476 nm的發(fā)光機理。實驗和理論計算結果證明分子間苯基的π–π空間共軛是該化合物固態(tài)發(fā)光的主要光物理作用機制[15]。
吸收波長紅移是產(chǎn)生共軛體系的一個典型特征。分子的平面化也能使吸收波長紅移,但平面化引起的紅移一般為20–30 nm,并且具有結構振動峰,而分子聚集引起的紅移則往往比較大,并且精細結構峰消失[16]。THPs具有很強的聚集誘導發(fā)光(aggregation-induced emission,AIE)特性。AIE特性是2001年香港科技大學唐本忠院士課題組發(fā)現(xiàn)并命名的一種獨特熒光特性,即,在溶液中不發(fā)光,但聚集時發(fā)光的熒光特性[17]。AIE克服了傳統(tǒng)的熒光化合物在稀溶液中具有很高的熒光效率,但在濃溶液中或聚集時熒光減弱甚至淬滅的缺點,在多種應用領域中發(fā)揮了其獨特的優(yōu)勢。目前已發(fā)現(xiàn)THPs可以用于活細胞內質網(wǎng)成像[18]、可擦寫壓致熒光變色材料[19]以及靈敏的測定表面活性劑臨界膠束濃度的熒光點亮探針[20,21]等。THP-1b和1g還具有獨特的在不同溫度范圍的熒光溫度響應特性[22]。THPs的熒光特性不僅與其分子內的空間共軛有關,還與其分子間的電子相互作用有關。THP-1b和1g的激發(fā)波長比其在環(huán)己烷溶液中的吸收波長分別長38和92 nm,說明在THP-1b和1g中存在空間共軛。其在晶體結構中分子間芳基間的距離小于0.5 nm也證明了芳基π電子的相互作用[14],即存在空間共軛。
3.2 超共軛體系的結構特征
超共軛體系為σ鍵參與的共軛體系。自Mulliken[6]在1939年發(fā)現(xiàn)并提出碳氫σ鍵(σCH)參與的超共軛效應以來,超共軛體系得到了很大的發(fā)展[5,23,24]。除了傳統(tǒng)教材中提到的σCH外,其他多種原子與碳形成的σ鍵(σCX)也能參與共軛。超共軛體系的結構遠比共軛體系復雜。Alabugin和Manoharan[25]通過自然軌道(natural bond orbital,NBO)分析法計算了不同σCX鍵的供電子能力:(Al, Ga) >> Ge > As ≥Si > P > B > Se > H > C > S > Br > N > Cl > O > F (≥表示二者差別小于2.1 kJ˙mol?1,>>表示二者差別小于大于12.6 kJ·mol?1),并指出σCH與σCC鍵的供電子能力相差很微小,傳統(tǒng)上高估了二者的差別,該順序會隨分子結構不同而有所變化。Alabugin和Zeidan[26]通過NBO分析法計算的σCX鍵接受電子的能力順序為:Br (6.3) > Cl (6.2) > SH(1) (5.4) > F (5.1) > OH(1) (4.7) ≈ SH(2) (4.7) ≈ SeH (4.7) ≈ PH2(1)(4.6) ≈ AsH2(4.5) ≈ NH2(1) (4.5) > OH(2) (4.2) > PH2(2) (4.0) > NH2(2) (3.8) ≈ GeH3(3.8) > SiH3(3.6) >CH3(3.4) > H (3.2),這里的≈表示二者差別小于0.4 kJ·mol?1。
根據(jù)VB電子軌道類型,超共軛分為σ–π、σ–n和σ–σ三種(參見表1)。由于文獻在說明超共軛體系的結構特征及性質時常常以MO軌道示意圖加以說明。在下面闡明超共軛體系的實例中,除了用簡單明了的VB理論軌道示意圖(見圖1a,σ鍵和π鍵用原子軌道重疊示意圖表示)外,還同時給出MO軌道示意圖(見圖1b,成鍵分子軌道σ和π,以及反鍵分子軌道π*,用原子軌道重疊示意圖表示;而反鍵分子軌道σ*用分子軌道圖表示)。從MO理論來看,σCX鍵參與共軛時,即可作為電子供體,以σCX成鍵分子軌道參與共軛;又可作為電子受體,以σ*CX反鍵分子軌道參與共軛。那么σCX參與共軛時到底是作為電子供體,還是作為電子受體,取決于X原子的電負性以及軌道的電子云密度大小。在其他條件相同的情況下,與電負性較大的原子形成的σCX鍵作為電子受體的能力較大,而作為供體的能力較小;反之,則作為電子供體的能力較大,而作為受體的能力較小。
3.2.1σ-π超共軛體系結構特征
如圖8所示,σCX鍵以單鍵(標示紅色的單鍵)與雙鍵相連時,可以形成σ–π超共軛體系。傳統(tǒng)的有機化學教材只提到σCH–π超共軛,認為σCH能夠產(chǎn)生共軛效應的原因是因為氫原子小不能屏蔽σ鍵電子云,從而使σ鍵電子軌道可以與相鄰的π軌道重疊,形成超共軛。如圖8a所示,丙烯分子中甲基上的3個σCH鍵與C=C雙鍵之間有單鍵(標示紅色的單鍵)相連,可以形成σCH–π超共軛。當我們把甲基上的一個σCH鍵以及π鍵用電子軌道表示時,就可以很清楚地看到σCH鍵與π鍵的p道相鄰,即可以發(fā)生重疊(見圖8a中VB理論電子軌道示意圖)。但這種重疊是從側面發(fā)生的,其重疊程度遠比π–π、p–π共軛體系中電子軌道的重疊程度小。所以,超共軛體系中的電子離域程度比π–π、p–π共軛體系離域程度弱。從MO理論來看,此時的σCH鍵在共軛體系中是作為電子供體,以成鍵分子軌道σCH參與共軛,而π鍵則作為電子受體,以π*CC反鍵分子軌道參與共軛,形成σCH-π*CC超共軛(見圖8a中MO理論電子軌道示意圖)。σCH成鍵分子軌道與π*CC反鍵分子軌道相互作用形成能量更低的成鍵分子軌道和能量更高的反鍵分子軌道,兩個電子進入能量更低的成鍵分子軌道,體系能量降低(見圖8a中MO能級示意圖)。
除σCH鍵可以形成超共軛外,已報道其他原子與碳形成的σ鍵也可以形成超共軛。如圖8b所示,全氟代環(huán)丁烯醇和全氟代環(huán)戊烯醇分子中有四個碳氟σCF鍵與π鍵之間有單鍵(標示紅色的單鍵)相連,可以形成σCF–π超共軛。一般來說,由于環(huán)張力的存在,四元環(huán)的穩(wěn)定性比五元環(huán)的穩(wěn)定性差。但全氟代環(huán)丁烯醇的穩(wěn)定性卻比全氟代環(huán)戊烯醇的穩(wěn)定性大[4]。這是因為四元環(huán)中的σCF鍵與π鍵的空間距離比五元環(huán)中的σCF鍵與π鍵的空間距離更近(見圖8b中VB理論電子軌道示意圖),具有更大的重疊程度,即更大的共軛程度所致[4]。由于F原子電負性比較大,從MO理論來看,σCF鍵在共軛體系中作為電子受體,以σ*CF反鍵分子軌道參與共軛,而π鍵則作為電子供體,以πCC成鍵分子軌道參與共軛,形成πCC–σ*CF超共軛(見圖8b中MO理論電子軌道示意圖)。σ*CF反鍵分子軌道與πCC成鍵分子軌道相互作用形成能量更低和更高的兩個分子軌道,兩個電子進入能量更低的分子軌道,體系能量降低(見圖8b中MO能級示意圖)。

圖8 分子中存在σ-π超共軛體系的化合物
3.2.2σ-n超共軛體系結構特征
σ–n超共軛體系為σCX鍵與非鍵n電子軌道(非鍵p軌道和雜化軌道)重疊形成的共軛體系。如圖9a–d所示,乙基正離子(CH3CH2+)、乙基自由基(CH3CH2·)、2-氟代乙基負離子(CH2FCH2-)和氟代甲氨分子(FCH2NH2)分子中存在σ–n超共軛體系。從VB理論來看,這些分子中的σ–n超共軛體系的形成,是以單鍵(標示紅色的單鍵)相連的鄰位σCX鍵與n電子軌道相互重疊所致(見圖9a–d的第2行)。從MO理論來看,在乙基正離子和乙基自由基中,缺電子的碳正離子和碳自由基價層上分別含0個及1個電子的p軌道作為電子受體參與共軛,而含滿電子的成鍵分子軌道σCH作為電子供體參與共軛,形成σCH–p(空)或與σCH–p(單)超共軛(見圖9a和b的第3行)。p軌道與σCH成鍵分子軌道相互作用形成能量更低和更高的兩個分子軌道,電子先填入能量更低的分子軌道,然后再填入能量更高的分子軌道,填入更低能量分子軌道的電子數(shù)多于填入更高能量分子軌道的電子數(shù),體系能量降低(見圖9a和b的第4行)。但在2-氟代乙基負離子和氟代甲氨分子中,是含電負性大的F原子σCF鍵以反鍵分子軌道σ*CF作為電子受體參與共軛,而滿電子的p軌道或sp3雜化軌道作為電子供體參與共軛[5],形成σ*CF–p或與σ*CF–sp3超共軛(見圖9c和d的第3行)。σ*CF與p軌道或sp3雜化軌道相互作用形成能量更低和更高的兩個分子軌道,一對電子填入能量更低的分子軌道,體系能量降低(見圖9c和d的第4行)。

圖9 分子中含σ–n超共軛體系的化合物
在有機化學中,一般認為取代環(huán)己烷的穩(wěn)定構象是較大基團以e鍵相連的、空間位阻較小的椅式構象。但當有電負性較大的元素存在時,情況并非如此。如圖10a中的2-氯四氫-2H-吡喃的穩(wěn)定構象是氯原子位于a鍵位的椅式構象(I),而不是氯原子位于e鍵位的椅式構象(II),I式轉變成II式的自由能為9.08 kJ·mol?1[27]。這是因為I式中氧原子上含孤對電子的p軌道與反鍵分子軌道σ*CCl幾乎平行,重疊程度比較大,可以形成更低能量的分子軌道,即形成更穩(wěn)定的p–σ*CCl超共軛體系。這種現(xiàn)象被稱為異頭效應(anomeric effect),是Edward[28]于1955年首次在吡喃糖中發(fā)現(xiàn)的。σ–n超共軛對反應活性也有很大影響。如圖10b所示,由于σCSi比σCH具有更強的給電子能力,可以與碳正離子的空p軌道形成更穩(wěn)定的σ–n超共軛體系,使得僅僅將H換成SiCH3的消除反應速率增大2.4 ×1012倍[29]。

圖10 σ–n超共軛對分子結構及反應活性的影響
3.2.3σ-σ超共軛體系結構特征
σ–σ超共軛體系是指由于相鄰的σ鍵電子軌道的重疊而形成的超共軛體系。如圖11a所示,乙烷分子具有兩個典型的構象,即能量最高的重疊式和能量最低的交叉式。有關交叉式構象比重疊式構象穩(wěn)定的原因,有機化學教材上的解釋是重疊式中非鍵氫原子之間的距離(229 pm)小于兩個氫原子范德華半徑之和(240 pm)[9](圖11b)或者認為是重疊式中C―H鍵上的σ電子對的排斥力較大[30](圖11c)。1939年,Mulliken[6]首次指出超共軛效應是乙烷交叉式更穩(wěn)定的主要原因。2001年,Vojislava Pophristic和Lionel Goodman[31]指出交叉式構象中含電子的σCH成鍵分子軌道與空的σ*CH反成分子鍵軌道重疊形成了更穩(wěn)定的分子軌道,即形成了σCH–σ*CH共軛體系,從而使交叉式構象更穩(wěn)定(σCH–σ*CH軌道重疊圖如圖11d所示)。超共軛效應是乙烷交叉式更穩(wěn)定的主要原因得到了一些實驗及理論的支持[32]。

圖11 乙烷分子(CH3CH3)的交叉式(staggered)和重疊式(eclipsed)構象
前面已經(jīng)提到,從MO理論來看,σ鍵參與共軛時,與碳形成σ鍵的原子電負性越大,該σ鍵作為電子受體的能力越大,而所為電子供體的能力越弱;反之,則作為電子供體的能力越大,而所為電子受體的能力越弱。這就是說,當兩個電負性相差比較大的原子與碳形成的σ鍵相鄰時,電負性比較大的原子與碳形成的反鍵分子軌道σ*可以作為有利的電子受體(acceptor),用σ*CA表示,而電負性比較小的原子與碳形成的成鍵分子軌道σ可以作為有利的電子供體(donor),用σCD表示。σ*CA與σCD相互作用,會產(chǎn)生更強的σ–σ超共軛效應。如圖12a和b中1,2-二氟乙烷[33]和3-氟-N,N-二甲基哌啶正離子[34]的I式構象比II式構象能量更低更穩(wěn)定,以及圖12c中Z-1,2-二氟乙烯比E-1,2-二氟乙烯能量更低更穩(wěn)定[35]。這是因為,在能量更低的構象式或順反異構式中,σ*CA與σCD可以發(fā)生重疊(見圖12中第2行MO理論電子軌道示意圖,D原子用綠色表示,A原子用藍色表示),形成穩(wěn)定的超共軛體系。而在能量較高的構象式或順反異構式中,σ*CA與σCD的重疊很少,不能形成穩(wěn)定的超共軛體系。
3.2.4 不同位置的超共軛體系結構特征
前面討論的超共軛體系分類是基于軌道類型的分類。同樣,超共軛還可以根據(jù)相鄰的原子或基團之間是否有價鍵相連,把超共軛分為通過價鍵的超共軛和通過空間的超共軛。在無σ鍵參與的共軛體系中,價鍵共軛體系中的原子或原子團是由σ單鍵相連的鄰位原子或原子團。但在超共軛體系中,價鍵超共軛體系中的原子或原子團之間除了是由σ單鍵相連的鄰位原子或原子團外,還可以是以雙鍵相連的鄰位原子或基團(見圖12c)。另外,連接同一碳原子上的兩個原子或基團也可以形成超共軛。如圖13a所示,化合物3中連接同一碳上的兩個σCC單鍵可以形成超共軛,并且該化合物中存在的超共軛效應遠大于乙烷分子交叉構象中兩個σCH單鍵的超共軛效應(NBO法計算的σwing–σ*inner與σwing–σ*wing之間的軌道作用能分別為69.5 kJ·mol?1和83.3 kJ·mol?1,而乙烷分子中的σCH–σ*CH僅為12.1 kJ·mol?1)[36]。從13a中MO電子軌道示意圖可以看到,同碳原子上的鍵角α越小,σ-σ*的軌道重疊程度越大,即超共軛效應越大。這就是為什么化合物3中σwing–σ*inner與σwing–σ*wing兩種類型的超共軛效應比乙烷分子中σCH–σ*CH超共軛效應大的原因。

圖12 σ–σ共軛對分子結構的影響
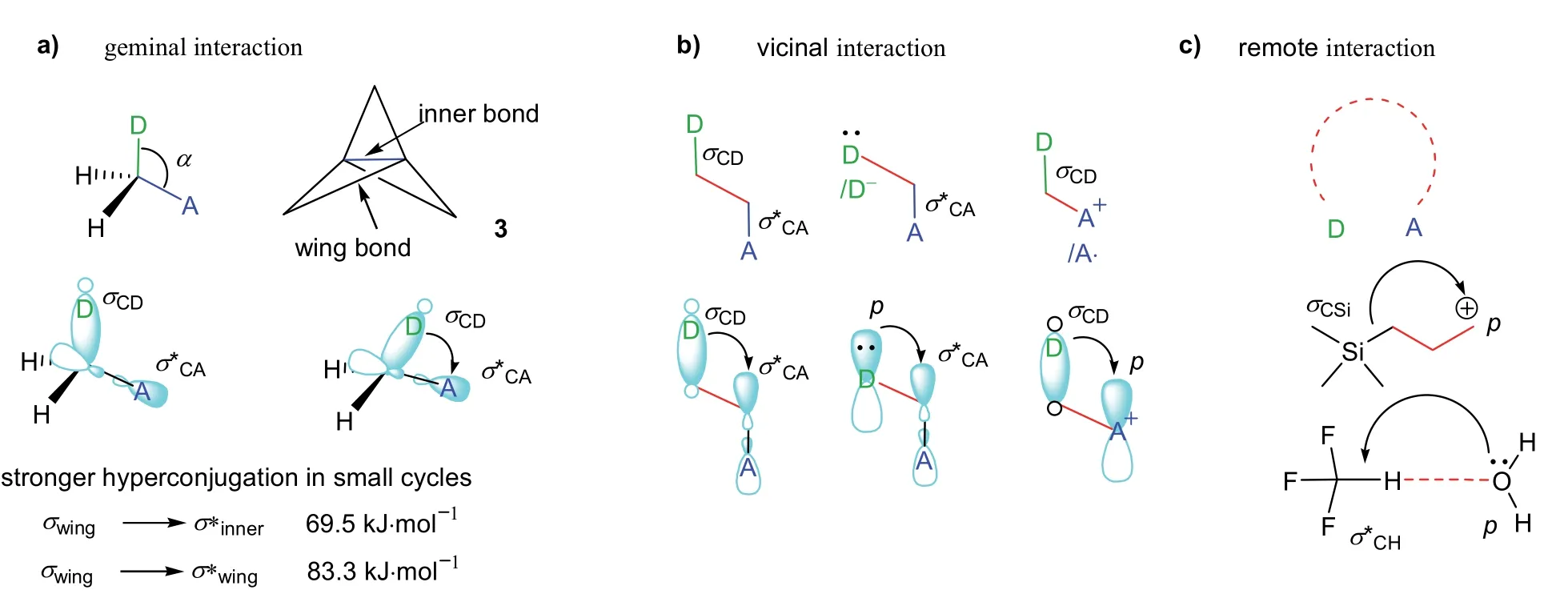
圖13 基于超共軛體系中原子或原子團之間連接方式的超共軛體系分類[5]
空間超共軛體系中的原子或原子團的相對位置也比前面的空間共軛體系復雜。根據(jù)形成超共軛體系的原子或原子團是連接同一碳原子上的原子或原子團、還是連接在單鍵或雙鍵兩端的鄰位原子或原子團、還是非直接相連的原子或原子團,Alabugin等[5]將超共軛體系分為同位(geminal)、鄰位(vicinal)和遠程(remote)原子或原子團軌道相互作用的超共軛體系(如圖13a–c所示)。這里的遠程超共軛類似前面討論的空間共軛,可以發(fā)生在分子內,也可以發(fā)生在分子之間。分子內的遠程超共軛體系可以是通過中間間隔多個價鍵的原子或基團。如圖13c所示,在3-(三甲基硅烷基)丙基正離子中相隔兩個σ鍵(標示紅色的兩個σ鍵)的碳正離子與C―Si基團發(fā)生σCSi―p超共軛[37]。遠程超共軛體系也可以是分子間相鄰的原子或基團。如圖13c所示的三氟甲烷中的C―H基團與水分子中的氧原子之間形成的σCH―p超共軛[38]。
4 結語
綜上所述,共軛體系是相鄰原子或基團的電子軌道發(fā)生重疊,電子發(fā)生離域運動,鍵長平均化,體系能量降低的體系,可以存在于分子內和分子間,對化合物的結構、物理與化學性質有很大的影響。從上面的實例可以看出,相鄰原子或基團的電子軌道重疊是共軛體系的本質特征。共軛體系可以根據(jù)電子軌道類型、相鄰基團是否有價鍵相連以及共軛效應作用強度進行分類。廣義的共軛體系包含所有電子軌道類型的共軛體系。但一般把無σ鍵參與的共軛體系稱為共軛體系,而把有σ鍵參與的共軛體系稱為超共軛體系。共軛和超共軛體系又可以根據(jù)參與共軛的軌道類型進行進一步分類(基于VB理論及MO理論中電子軌道的共軛體系分類見表1)。共軛和超共軛體系還可以根據(jù)共軛體系中相鄰的原子或基團是否有價鍵相連,把共軛和超共軛體系分為價鍵共軛與空間共軛。空間共軛和超共軛又可分為分子內與分子間的空間共軛。一般文獻或教材只是用VB或者MO理論的電子軌道圖闡明共軛體系的分類及結構特征。本文通過實例,同時用VB和MO理論的電子軌道示意圖闡明復雜的超共軛體系分類及結構特征,有助于從不同角度加深對共軛體系結構特征及共軛效應本質的理解。
猜你喜歡
《學習方法報》歷史中考版(2023年21期)2023-11-09 07:40:38
數(shù)學小靈通(1-2年級)(2020年9期)2020-10-27 03:24:18
當代貴州(2019年41期)2019-12-13 09:28:56
娃娃樂園·3-7歲綜合智能(2016年1期)2016-10-25 09:32:48
中國共青團(2015年7期)2015-12-17 01:24:38
中學生數(shù)理化·八年級物理人教版(2014年1期)2015-01-09 08:50:45
中國扶貧(2014年8期)2014-06-27 15:33:39
中國扶貧(2014年8期)2014-06-27 04:09:02
中學生數(shù)理化·八年級物理人教版(2014年2期)2014-04-02 08:50:44
能源(2014年3期)2014-03-27 09:55:20
