(2R,3R)-1,4-二甲氧基-1,1,4,4-四苯基-2,3-丁二醇合成方法研究進(jìn)展
2021-06-03 07:25:08胡曉允單自興
大學(xué)化學(xué) 2021年4期
胡曉允,單自興
1中南民族大學(xué)化學(xué)與材料科學(xué)學(xué)院,武漢 430074
2武漢大學(xué)化學(xué)與分子科學(xué)學(xué)院,武漢 430072
(2R,3R)-1,4-二甲氧基-1,1,4,4-四苯基-2,3-丁二醇(1)是天然酒石酸衍生的一種 C2-對(duì)稱的位阻型手性二醇,在有機(jī)合成中有著廣泛的應(yīng)用。(2R,3R)-1作為手性助劑在硼酸手性化學(xué)中的研究最為深入,如作為手性硼酸的保護(hù)基團(tuán)用于不對(duì)稱環(huán)丙烷化、環(huán)氧化、3,3-Sigmatropic重排和羰基的烯丙基化反應(yīng)。2012年,Pietruszka等[1]就(2R,3R)-1的制備和應(yīng)用專門撰寫了一篇綜述。
盡管(2R,3R)-1已經(jīng)發(fā)展為一類應(yīng)用廣泛的手性二醇,但其制備方法仍停留在30年前的合成方案,關(guān)于它的合成研究報(bào)道較少。最早報(bào)道(2R,3R)-1的合成方案是從天然酒石酸酸酯出發(fā),經(jīng)5步反應(yīng)制得,其關(guān)鍵步驟是光學(xué)純酒石酸酯仲羥基的保護(hù)及去保護(hù)和叔羥基的甲基化程序:仲羥基的去保護(hù)涉及到2,3-二氯-5,6-二氰基苯醌(DDQ)氧化和LiAlH4還原程序,而叔羥基的甲基化則需要利用NaH/MeI等試劑[2]。2009年開始,本課題組對(duì)(2R,3R)-1,1,4,4-四取代丁四醇的制備及區(qū)域選擇性轉(zhuǎn)變化學(xué)進(jìn)行了持續(xù)的研究,基于(2R,3R)-1,1,4,4-四苯基丁四醇(2)的區(qū)域選擇性 2,3-螺硼化反應(yīng)(硼化學(xué)保護(hù))和 2,3-亞硫酸酯化反應(yīng)(環(huán)亞硫酸酯保護(hù)),相繼發(fā)展了兩種方案制備(2R,3R)-1。前者避免了DDQ-LiAlH4氧化-還原去保護(hù)程序,但仍采用NaH/MeI體系進(jìn)行甲基化,且利用HF進(jìn)行水解脫硼,在制備成本和環(huán)境保護(hù)方面存在一些問題。環(huán)亞硫酸酯保護(hù)方案利用常規(guī)試劑甲醇引入甲氧基,避免了NaH/MeI試劑的使用;另一方面利用無機(jī)堿液進(jìn)行去保護(hù),取代了DDQ-LiAlH4氧化-還原去保護(hù)程序。相對(duì)于原合成方案,本課題組發(fā)展的合成方案縮短了合成路線,降低了制備成本,具有高效、便捷、綠色化程度較高的優(yōu)勢(shì)。本文主要對(duì)(2R,3R)-1合成方法的研究進(jìn)展進(jìn)行概述。
1 縮醛保護(hù)法制備(2R,3R)-1
1.1 Nakayama合成路線
(2R,3R)-1的合成路線最初是由Nakayama和Rainier[2]在1990年提出來。如圖1所示,天然酒石酸甲酯與對(duì)甲氧基苯甲醛的縮醛化合物在對(duì)甲苯磺酸催化下發(fā)生醇置換,生成的縮醛將酒石酸酯2,3-位仲羥基保護(hù)起來,繼而與苯基格氏試劑反應(yīng),對(duì)酯基進(jìn)行烴基化生成手性二醇。接下來,利用NaH/MeI對(duì)新生成的兩個(gè)叔羥基進(jìn)行醚化。最后,用DDQ進(jìn)行氧化,得到羥基酯,再用LiAlH4還原將兩個(gè)仲羥基游離出來,通過5步反應(yīng)以37%的總收率得到(2R,3R)-1。
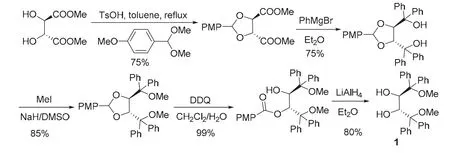
圖1 Nakayama合成路線[2]
縮醛在酸性條件下易水解得到相應(yīng)的醛和醇。但 Nakayama合成方案中,形成的縮醛穩(wěn)定性較好,且酸性條件下,新生成的甲氧基易被水解。所以采用DDQ-LiAlH4進(jìn)行氧化-還原將2,3-位仲羥基游離出來。這無疑增加了制備(2R,3R)-1的成本和合成操作的繁瑣,尤其 LiAlH4的使用需要無水操作,大量使用具有一定的危險(xiǎn)性。
1.2 Nakayama合成路線的改進(jìn)
2008年,Pietruszka研究組[3]對(duì)該合成方案進(jìn)行了改進(jìn)。如圖2所示,采用便宜的無機(jī)鹽溴酸鈉和連二亞硫酸鈉代替DDQ對(duì)縮醛進(jìn)行氧化,但仍需用LiAlH4進(jìn)行還原,其他步驟和Nakayama方案一致。出乎意料的是,該合成方案中,利用苯基格氏試劑對(duì)酯基進(jìn)行烴基化時(shí),Pietruszka采用甲基四氫呋喃代替乙醚做溶劑,產(chǎn)率由75%提高到98%,反應(yīng)總收率提高到48%。
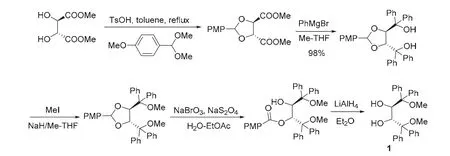
圖2 Pietruszka研究組合成路線[3]
2 (2R,3R)-2的區(qū)域選擇性轉(zhuǎn)變制備(2R,3R)-1
(2R,3R)-2 是 TADDOL (α,α,α’,α’-tetraaryl-2,2-dimethyl-1,3-dioxolan-4,5-dimethanol)母體化合物,盡管TADDOL是一種通用的手性二醇,在不對(duì)稱合成中應(yīng)用廣泛[4,5],但關(guān)于其母體化合物(2R,3R)-2的研究卻很少。2010年,本課題組嘗試通過硼化學(xué)方法將酒石酸酯的仲羥基保護(hù)起來,如圖3所示,最初設(shè)計(jì)合成路線Route A:通過苯基硼酸將酒石酸二乙酯2,3-仲羥基保護(hù)起來,繼而與苯基格氏試劑反應(yīng),再水解脫去硼保護(hù)基得到(2R,3R)-2。考慮到苯基硼酸一般通過硼酸酯與苯基格氏試劑制備,其成本相對(duì)比較高昂,又設(shè)計(jì)合成路線Route B:硼酸三丁酯代替苯基硼酸對(duì)酒石酸酯的仲羥基進(jìn)行保護(hù)。這兩種方案制備(2R,3R)-2的產(chǎn)率基本相當(dāng),但第二條路線無疑成本要低廉些。上面兩條合成路線相對(duì)于傳統(tǒng)的縮醛保護(hù)和去保護(hù)方法成本低廉、操作方便,但還需要保護(hù)和去保護(hù)程序,且浪費(fèi)了硼試劑。為了進(jìn)一步簡(jiǎn)化實(shí)驗(yàn)步驟,提高原子經(jīng)濟(jì)性,我們?cè)O(shè)計(jì)合成路線Route C:苯基格氏試劑直接與酒石酸二乙酯“一鍋”反應(yīng)制備(2R,3R)-2。盡管該合成方案浪費(fèi)了格氏試劑,但縮短了反應(yīng)的操作程序,這三條合成路線以基本相當(dāng)?shù)漠a(chǎn)率得到目標(biāo)化合物。因此,我們選定酒石酸酯與格氏試劑“一鍋”法合成了一系列(2R,3R)-1,1,4,4-四取代丁四醇[6]。
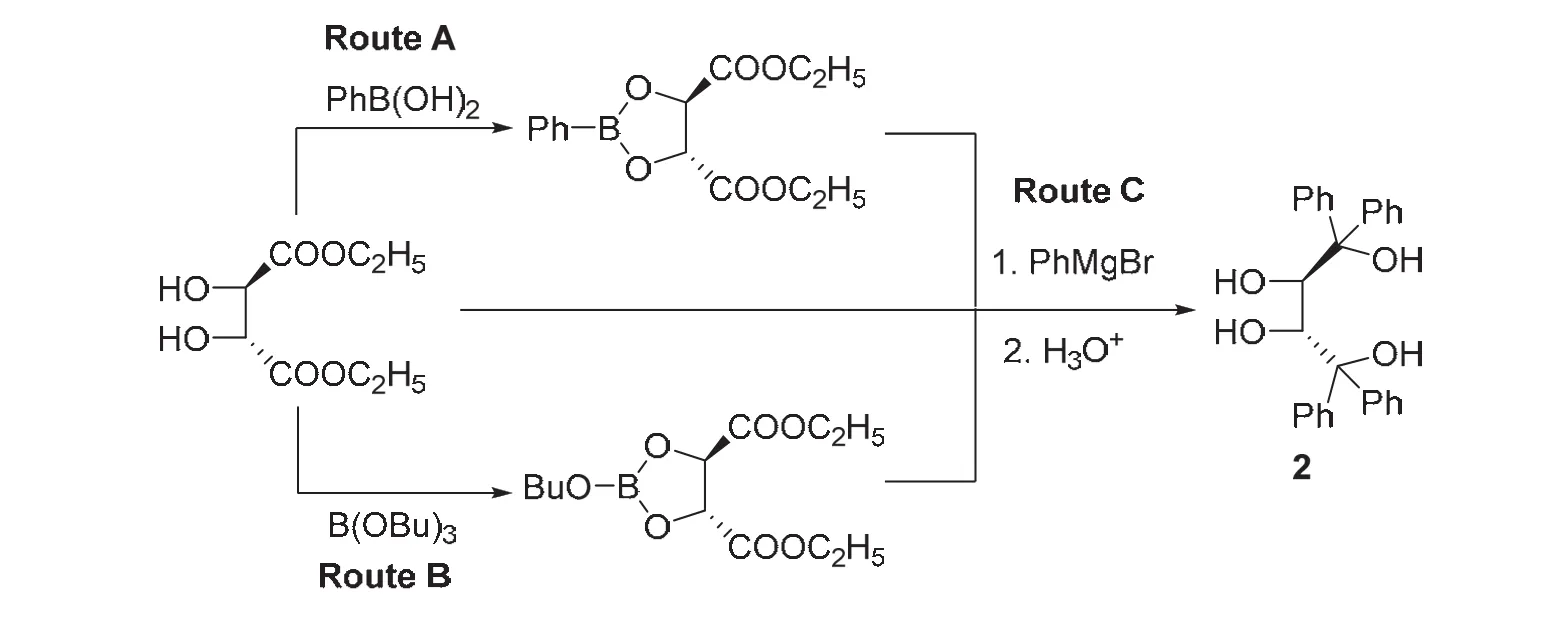
圖3 (2R,3R)-2的合成路線[6]
2.1 (2R,3R)-2硼化學(xué)保護(hù)法合成(2R,3R)-1
(2R,3R)-2有兩個(gè)仲羥基和兩個(gè)叔羥基,叔羥基所連碳原子上有苯基取代基,空間位阻和電子性質(zhì)決定仲羥基和叔羥基應(yīng)具有不同的反應(yīng)行為。我們?cè)O(shè)想利用仲羥基和叔羥基反應(yīng)活性的差異及苯基取代基的空間位阻對(duì)其實(shí)現(xiàn)選擇性衍生化,避開縮醛保護(hù)和去保護(hù)的繁瑣步驟。經(jīng)過探索,我們發(fā)現(xiàn)(2R,3R)-2與不同的硼試劑反應(yīng)時(shí),會(huì)表現(xiàn)出不同的區(qū)域選擇性。如圖4所示,(2R,3R)-2與烴基硼酸反應(yīng)時(shí),會(huì)發(fā)生高度區(qū)域選擇性 1,3-環(huán)硼化反應(yīng),生成手性雙環(huán)[4.4.0]二硼酸酯(3)[7];但在堿性條件下,與硼酸則發(fā)生高度區(qū)域選擇性2,3-螺硼化反應(yīng)生成手性螺硼酸鹽(4)[8]。
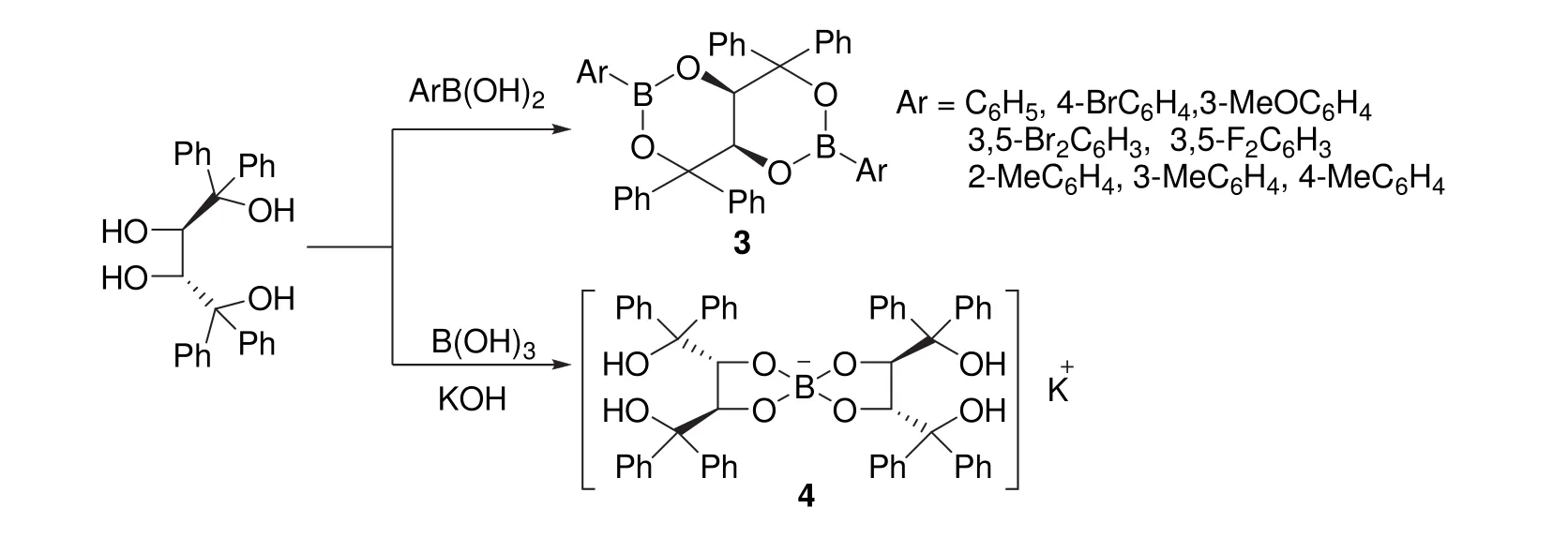
圖4 (2R,3R)-2與硼試劑的區(qū)域選擇性衍生化[7,8]
手性螺硼酸鹽4性質(zhì)很穩(wěn)定,且容易制備,通過一個(gè)硼原子將兩分子(2R,3R)-2的仲羥基選擇性地保護(hù)起來。利用這一特性,我們?cè)O(shè)計(jì)如下合成路線制備(2R,3R)-1。如圖5所示,天然酒石酸二乙酯與苯基格氏試劑“一鍋”反應(yīng)制得(2R,3R)-2,在強(qiáng)堿KOH溶液存在下,(2R,3R)-2與硼酸發(fā)生高度區(qū)域選擇性 2,3-螺硼化反應(yīng),近乎定量地得到手性螺硼酸鉀鹽 4。接下來,通過傳統(tǒng)的 NaH/MeI甲基化程序?qū)κ中月菖鹚猁}4的叔羥基進(jìn)行醚化,最后用HF的甲醇溶液進(jìn)行去保護(hù),將兩個(gè)仲羥基游離出來,高效地制得(2R,3R)-1。該合成方案用廉價(jià)的硼酸和無機(jī)酸堿取代了DDQ和LiAlH4等試劑,簡(jiǎn)化了合成方案,將報(bào)道的5步合成反應(yīng)優(yōu)化至4步,反應(yīng)總收率達(dá)到45%[8]。
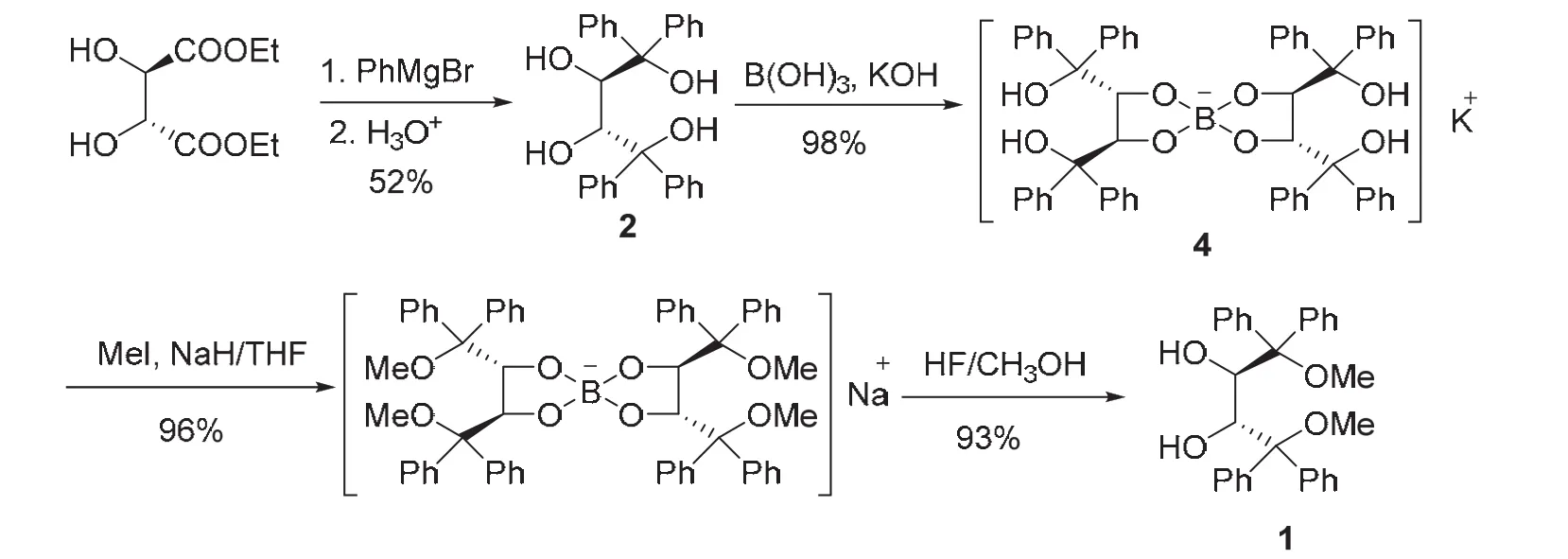
圖5 硼化學(xué)保護(hù)法合成路線[8]
盡管該合成方案提高了原子利用率,簡(jiǎn)化了合成方案,但仍然存在以下兩點(diǎn)不足:1) 1,4-位甲氧基的引入采用傳統(tǒng)的醚化試劑MeI/NaH,制備成本仍然較高,工業(yè)生產(chǎn)一般用硫酸二甲酯做甲基化試劑,但硫酸二甲酯和MeI均為毒性較大的試劑;2) 反應(yīng)最后切斷B―O鍵需要使用HF。HF不僅毒性較大,還具有極強(qiáng)的腐蝕性,能強(qiáng)烈地腐蝕金屬、玻璃和含硅的物體,一般需要在塑料容器中開展實(shí)驗(yàn)。所以從綠色化學(xué)的角度審視該合成方案,仍有繼續(xù)優(yōu)化的空間。
2.2 (2R,3R)-2環(huán)亞硫酸酯保護(hù)法合成(2R,3R)-1
我們?cè)诳疾?2R,3R)-2的反應(yīng)化學(xué)時(shí),發(fā)現(xiàn)在一定條件下,其能與氯化亞砜發(fā)生高度區(qū)域選擇性2,3-環(huán)亞硫酸酯化反應(yīng),生成雙羥基環(huán)亞硫酸酯(4R,5R)-5,游離的叔羥基與氯化亞砜進(jìn)一步反應(yīng)可以被氯代,得到二氯代環(huán)亞硫酸酯(4R,5R)-6。研究發(fā)現(xiàn),控制氯化亞砜的用量,也可以實(shí)現(xiàn)(2R,3R)-2與氯化亞砜一步反應(yīng)直接制得(4R,5R)-6 (圖6)[9]。
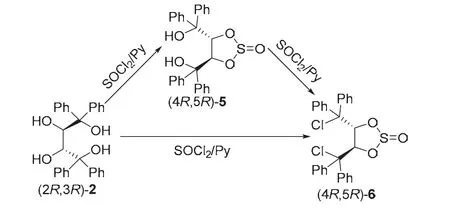
圖6 (2R,3R)-2與氯化亞砜的區(qū)域選擇性環(huán)亞硫酸酯化反應(yīng)[9]
我們知道,鹵代烴在有機(jī)合成中是關(guān)鍵的合成中間體,親核取代反應(yīng)是其典型的化學(xué)性質(zhì)之一,C―Cl極性鍵易于被各種親核試劑進(jìn)攻,引入相應(yīng)的官能團(tuán)。我們?cè)O(shè)想利用甲醇作為親核試劑與二氯代環(huán)亞硫酸酯(4R,5R)-6反應(yīng),引入甲氧基,從而避開羥基的甲基化程序。基于這一設(shè)想,設(shè)計(jì)了如下合成方案,如圖7所示,天然酒石酸酯二乙酯經(jīng)過烴基化反應(yīng)制得(2R,3R)-2,與氯化亞砜發(fā)生高度區(qū)域選擇性2,3-環(huán)亞硫酸酯化反應(yīng)生成(4R,5R)-6,與甲醇發(fā)生親核取代反應(yīng)引入甲氧基,形成雙甲氧基環(huán)亞硫酸酯(4R,5R)-7,用堿液處理脫去亞硫酰基,經(jīng)4步反應(yīng),以45%的總收率制得(2R,3R)-1[9]。
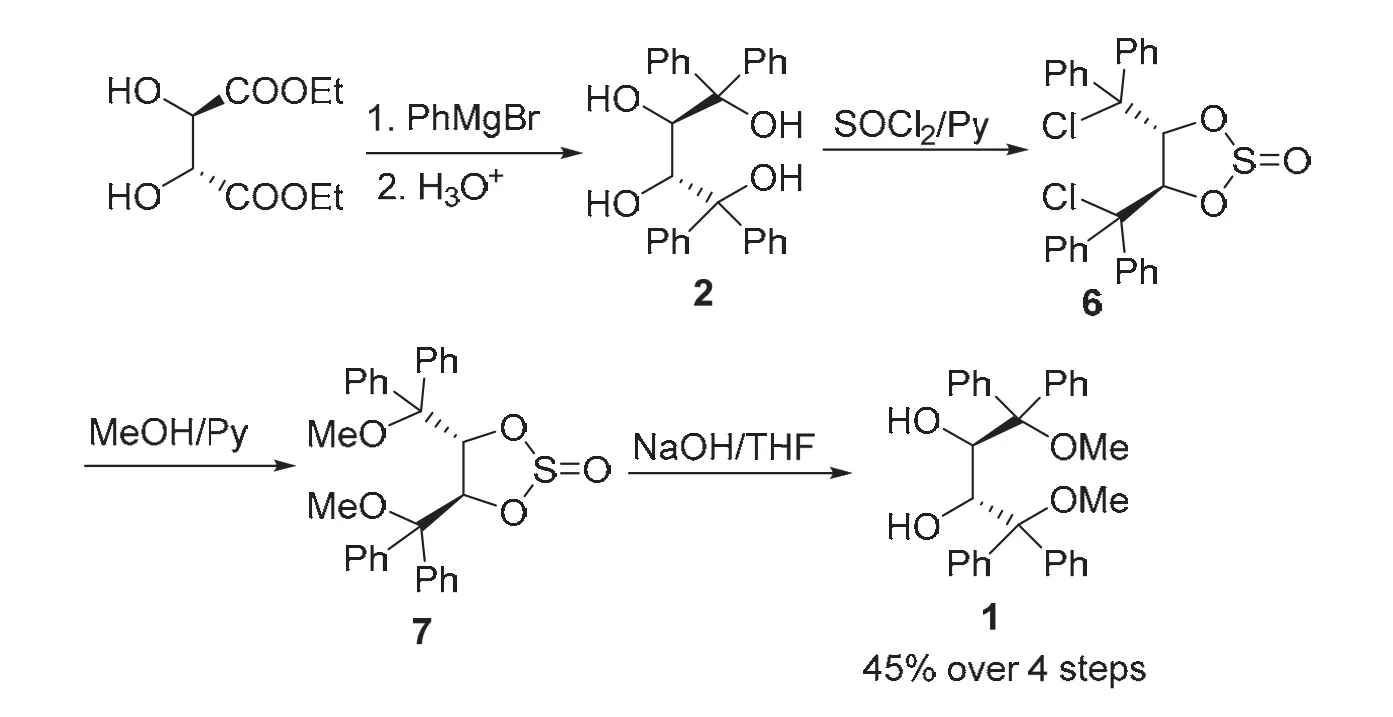
圖7 環(huán)亞硫酸酯保護(hù)法合成路線[9]
該合成方案利用亞硫酰基實(shí)現(xiàn)對(duì)酒石酸酯仲羥基的保護(hù),而其去保護(hù)通過常規(guī)堿液即可實(shí)現(xiàn),相對(duì)于縮醛和螺硼酸鹽的去保護(hù)方法,該方法高效便捷,易于大規(guī)模制備;另一方面,利用甲醇與鹵代烴的親核取代反應(yīng)引入甲氧基,避開了通過MeI/NaH對(duì)羥基醚化的方案,不僅大大降低了制備成本,簡(jiǎn)化了實(shí)驗(yàn)操作步驟,而且避免了毒性較大的甲基化試劑的使用,提高了該合成方案的綠色化程度。
旋光測(cè)定揭示在通過(2R,3R)-2的區(qū)域選擇性2,3-環(huán)亞硫酸酯化制備(2R,3R)-1過程中,即使用強(qiáng)堿氫氧化鈉溶液處理,手性中心也未發(fā)生消旋化。由此推測(cè)在環(huán)亞硫酸酯水解過程中,斷鍵反應(yīng)未涉及到手性中心。基于該實(shí)驗(yàn)結(jié)果,提出環(huán)亞硫酸酯(4R,5R)-7水解機(jī)理如圖8所示:OH?首先進(jìn)攻環(huán)亞硫酸酯的硫原子,而非手性碳原子。亞硫酰基斷鍵,接下來S-O鍵斷裂,新生成的O?奪取水中的H質(zhì)子形成羥基,OH?繼續(xù)進(jìn)攻硫原子,再斷去另一個(gè)S-O鍵,從而游離出兩個(gè)仲羥基[9]。
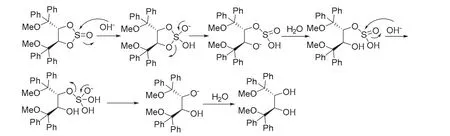
圖8 環(huán)亞硫酸酯(4R,5R)-7可能的水解機(jī)理[9]
3 結(jié)語
(2R,3R)-1合成方法的不斷優(yōu)化,是從綠色化學(xué)的角度去審視傳統(tǒng)的合成方案,通過高度區(qū)域選擇性控制,發(fā)展出高效、便捷、綠色化程度較高的合成方案。以綠色化學(xué)理念審視已有的合成方案,開發(fā)具有高度選擇性,包括化學(xué)選擇性、區(qū)域選擇性及立體選擇性的合成路線,提高合成方案的原子經(jīng)濟(jì)性,這在有機(jī)合成化學(xué)中仍極具發(fā)展空間。
