以不自主抖動為首發表現的脊髓小腦性共濟失調13型1病例報告
2021-05-25 01:58:30李旭穎王憲玲
中風與神經疾病雜志 2021年4期
關鍵詞:功能
郝 靜,李旭穎,王憲玲
常染色體顯性遺傳性小腦性共濟失調(ADCA)也稱為脊髓小腦性共濟失調(Spinocerebellar ataxia type,SCA),是一種起病隱匿,緩慢加重的神經系統變性病。其臨床表現非常廣泛,包括小腦性共濟失調、眼動障礙、錐體外系運動障礙、視神經萎縮、視網膜病變、認知障礙、周圍神經病及癲癇等[1]。由于各亞型間存在著巨大的重疊,臨床上對于特定亞型的診斷非常困難。隨著基因檢測技術的發展與應用,目前已鑒定出40余種亞型。SCA13型是臨床上罕見的一種類型,由于缺乏足夠的臨床數據,SCA13型的發病率尚不清楚。SCA13型在中國人群中罕見[2],此前國內僅報道過1個家系[3],該家系中發現了KCNC3基因未被文獻報道的新的突變位點c.1018G>A。現將我們發現的另1例SCA13型的病例報道如下,在該病例中以不自主抖動為首發表現,既往尚未見過類似報道。
1 病例資料
患者,男性,17歲,慢性起病,主因“雙手抖動10余年,言語不清、行走不穩7 y。”以“行走不穩待查”收入院。患者10余年前無明顯誘因出現雙手不自主抖動,表現為取物時抖動明顯,安靜時也可出現。雙手活動欠靈活,可完成日常生活。無明顯加重緩解因素。7 y前無明顯誘因出現言語不清,語速減慢,說話斷續。同時出現行走不穩,左右搖晃,無法走直線。未出現跌倒。偶有飲水嗆咳。入院查體:血壓:115/79 mmH,心率80次/分,呼吸20次/分,體溫36.5 ℃。神清,言語緩慢,粗測高級皮質功能基本正常。雙側瞳孔等大正圓,直徑約3.0 mm,對光反射靈敏。雙側眼球活動正常,雙側鼻唇溝對稱,伸舌居中,咽反射存在,余顱神經查體未見明顯異常。雙手可見不自主抖動。四肢肌力5級,雙下肢肌張力略增高。雙下肢腱反射(),雙側胸大肌反射陽性,雙側hoffmann征(-),雙側巴氏征(-)。深淺感覺查體未見異常。雙側指鼻,跟膝脛試驗欠穩準,輪替試驗欠靈活,閉目難立征陰性。一字步不能。腦膜刺激征陰性。個人史:無難產窒息病史,生長發育史正常。家族史:家族中未有類似疾病史。
輔助檢查:頭部磁共振成像(MRI)(見圖1):雙側小腦半球腦溝増寬、加深,小腦萎縮,左側顳角擴大。核磁共振波譜圖(MRS):右側小腦感興趣區NAA濃度(NAA/Cr:1.04),CHo濃度(CHo/Cr:0.97);左側小腦感興趣區NAA濃度(NAA/Cr:0.94),CHo濃度(CHo/Cr:1.01)。眼科檢查未見KF環。腰穿壓力 175 mmH2O,腦脊液白細胞計數1×106/L,腦脊液生化:腦脊液蛋白13 mg/dl,涂片找抗酸桿菌、細菌、真菌、墨汁染色、TORCH病毒檢測、24 h CSF IgG鞘內合成率及寡克隆帶均未見異常,自身免疫性腦炎、副腫瘤抗體:陰性。MMSE(簡易精神狀態檢查量表)評分:26分(學歷:初中)。MOCA(蒙特利爾認知評估量表):20分。畫鐘實驗1分。余常規化驗未見異常。
在本例中,排除常見的CAG重復序列導致的SCA亞型后,對患者及其父親進行全外顯子測序(母親拒絕檢測)。分析結果提示,患者的KCNC3基因檢測出雜合突變,父親為野生型,但由于缺少患者母親的基因信息,目前尚不能確定該基因突變是來源于母親還是新發突變。在本例中,該基因發現一處已報道的致病錯義突變c.1268G>A,導致第2號外顯子(總共5個外顯子)423位點上的精氨酸被組氨酸取代(R423H突變),且功能預測為臨床有害。由于:(1)KCNC3基因是SCA13的唯一致病基因;(2)該突變已有文獻報道,在群體中的攜帶頻率極低,功能預測為臨床有害突變,且遺傳模式符合常染色體顯性遺傳,ACMG預測為致病變異(PAT,PM1+PM2+PP3+PP2+PP5);(3)功能研究結果提示:KCNC3編碼電壓門控鉀離子通道蛋白3(Kv3.3)。該基因在腦中高表達,尤其在小腦,從遺傳和機制上能夠解釋本病例。進行一代測序驗證(見圖2),結果證實了突變的真實存在,符合SCA13型。在治療上主要給予B族維生素(維生素B1、維生素B6、甲鈷胺)營養神經等治療。半年后隨訪未進一步進展。
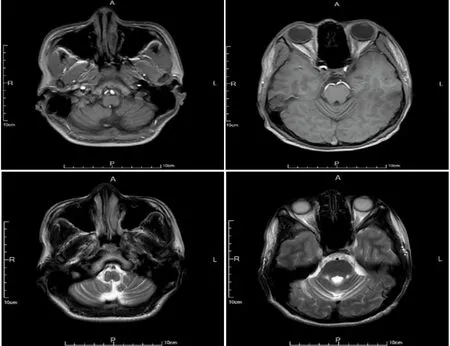
圖1 患者頭部MRI:可見雙側小腦半球腦溝増寬、加深
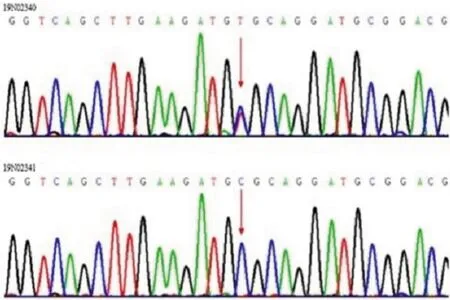
圖2 患者(上)及其父(下)基因檢測圖譜
2 討 論
SCA是遺傳性共濟失調類疾病的主要類型。一項納入了1062篇文章的薈萃分析發現,在SCA的患者中非共濟失調類的臨床表型也很常見[4]。該患者首次以不自主抖動為首發癥狀,該臨床表型尚未在SCA13型患者中進行過報道,容易造成在疾病早期誤診的可能。其次,該患者在安靜狀態及活動時均可見雙手非對稱性的抖動,接近物體時抖動無明顯加重(未行震顫分析),不符合典型的小腦性震顫。結合患者的發病年齡,首先需要除外其他運動障礙性疾病。不自主抖動屬于運動障礙的范疇,運動障礙是SCA重復擴增型患者的常見癥狀。高度提示在不同的亞型間臨床表型存在著巨大重疊。頭部MRI是SCA患者首選的影像學檢查。隨著疾病的進展,脊髓小腦性共濟失調的影像學可表現為脊髓萎縮、橄欖橋小腦萎縮和皮質小腦萎縮。在該患者中,可觀察到明顯的雙側小腦半球的萎縮,符合SCA的影像學表現。
相對于常見的SCA亞型,SCA13型的發病機制研究較少,基因KCNC3(也稱為Kv3.3)是SCA13型的唯一致病基因,位于染色體19q13.3-19q13.4,編碼Kv3.3電壓門控鉀通道。該通道主要分布在小腦浦肯野細胞、深部神經元、顆粒細胞等[5]。KCNC3基因的突變可能會導致Kv3.3通道蛋白功能改變,影響小腦的抗氧化應激的能力。該基因突變已在非洲爪蟾卵母細胞中得到驗證[6],SCA13型突變體主要影響了小腦神經元興奮性以及KCNC3表達的其他區域的神經元功能。目前在斑馬魚[7]、小鼠模型[8]中也進行了相關突變研究。在R424H突變的小鼠模型中,突變的結果會導致鈣通道的改變,影響神經元興奮性并導致小腦神經元的死亡,因此在SCA13型的患者中可觀察到明顯小腦萎縮。
既往已有文獻報道,證實R423H為致病性突變[9]。可有研究表明,SCA13型發病早晚和臨床癥狀可能與Kv3.3的激活轉移的方向有關[10]。R423H突變體可以抑制Kv3.3電流幅度,還可以顯著改變通道門控功能[9]。F448L和R423H突變體對通道門控有相似的作用[11],F448L會在超極化方向上改變通道激活電壓的依賴性,減慢通道關閉的速度。近年來,Duarri等人驗證了一個新的突變體V535M以及兩個可能的突變體D129N和S391G[10]。其中V535M和D129N也可以將Kv3.3通道激活轉移到超極化電壓。這4個突變體均改變了通道門控的功能,都表現為早期發病。R420H突變的患者臨床上表現為成年起病。經過功能驗證,R420H和R423H突變均位于Kv3.3通道S4跨膜區段中,均可產生顯性負性的功能亞基,但是R420H并未導致Kv3.3門控功能發生改變[11]。因此,發病年齡的早晚在很大程度上取決于通道門控功能的改變。同時有研究發現Kv3.3通道激活的去極化可能與痙攣性共濟失調步態有關[10]。此次報道的R423H突變的患者以錐體外系癥狀為首發表現。運動障礙考慮與基底節的神經元變性相關。震顫、肌張力障礙等運動障礙在SCA經典突變亞型中少見,該病例提示包括震顫在內的運動障礙似乎也是SCA13型臨床譜系的一部分,擴大了SCA13型的臨床表型庫,同時也在一定程度上支持了基因型-臨床表型的關聯。
針對SCA的治療,目前尚無特效的辦法來阻止疾病的進程[1]。國外利用慢性丘腦刺激治療了1例SCA2型患者的震顫[12]。在該患者中,在藥物治療方面主要給予了營養神經的B族維生素治療,并未找到特效的療法來延緩疾病發展。由于不同的亞型其發病機制存在一定的差異,已有試驗針對特定的亞型得到了某些有意義的結果。例如服用乙酰唑胺改善了SCA6型患者的共濟失調的癥狀[13]。目前有關SCA的研究還不夠充分,但是新的技術如蛋白組學分析等可幫助更好的尋找藥物對應的治療靶點,從而為尋找新的治療方向提供可能。
對于脊髓小腦性共濟失調,目前研究有限,在臨床上對于有共濟失調癥狀的患者,一定要追問其家族史,必要時行基因檢查。以錐體外系為首發表現的共濟失調的患者,應該考慮到該病的可能。臨床上需要多多留意相關患者,為進一步深入了解疾病提供幫助。
猜你喜歡
鐘表(2023年5期)2023-10-27 04:20:44
中華詩詞(2022年6期)2022-12-31 06:41:24
當代陜西(2021年21期)2022-01-19 02:00:26
中學生數理化(高中版.高考數學)(2020年1期)2020-02-20 13:23:44
經濟技術協作信息(2018年11期)2019-01-14 03:07:20
中國科技論壇(2017年7期)2017-07-25 08:49:53
制造技術與機床(2017年3期)2017-06-23 08:11:33
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
