豬肉中常見(jiàn)的9種蓄水類(lèi)藥物殘留的快速篩查
2021-05-14 00:30:54陳樹(shù)兵余曉玲方科益余曉琴王傳現(xiàn)
分析科學(xué)學(xué)報(bào) 2021年2期
陳樹(shù)兵,余曉玲,李 雙,方科益,余曉琴,韓 超,王傳現(xiàn)
(1.寧波海關(guān)技術(shù)中心,浙江寧波 315040;2.四川省食品藥品檢驗(yàn)檢測(cè)院,四川成都,610097;3.浙江樹(shù)人大學(xué)生物與環(huán)境工程學(xué)院,浙江杭州 310015;4.上海海關(guān)動(dòng)植物與食品檢驗(yàn)檢疫技術(shù)中心,上海 200135)
我國(guó)作為豬肉消費(fèi)大國(guó),其肉制品的生產(chǎn)安全直接關(guān)系到民生建設(shè)。近些年,出現(xiàn)的“注水肉”問(wèn)題與非法使用新型蓄水類(lèi)藥物直接相關(guān)。新型蓄水類(lèi)藥物主要包括:阻斷M膽堿受體的抗膽堿藥物(阿托品、東莨菪堿、山莨菪堿),局部麻醉藥物(普魯卡因、利多卡因),抗組胺類(lèi)藥物(異丙嗪)和腎上腺素受體激動(dòng)藥物(腎上腺素及其代謝物4-羥基-3-甲氧基-扁桃酸和3,4-二羥基扁桃酸)[1 - 10]。這些藥物的非法使用將不可避免地殘留于豬肉食品中,進(jìn)而對(duì)人體中樞神經(jīng)造成不良影響。目前,我國(guó)蓄水類(lèi)藥物殘留檢測(cè)標(biāo)準(zhǔn)缺乏,已有文獻(xiàn)多是按照某一類(lèi)藥物建立的,適應(yīng)范圍單一[1,2,8,11,12],尚無(wú)同時(shí)測(cè)定豬肉中9種蓄水類(lèi)藥物及其代謝物的報(bào)道。因此,建立豬肉中蓄水類(lèi)藥物的高通量篩查方法,對(duì)于提高監(jiān)管工作效率、保障豬肉食品安全具有重要的意義。
蓄水類(lèi)藥物檢測(cè)主要涉及前處理技術(shù)和儀器分析兩方面。已有方法主要采用固相萃取柱凈化。例如:王海燕等[1]對(duì)畜肉中阿托品類(lèi)藥物殘留量測(cè)定,前處理采用MCX小柱凈化;胡海山等[8]對(duì)動(dòng)物源性食品中鎮(zhèn)靜劑類(lèi)藥物殘留量測(cè)定,乙腈提取后經(jīng)Oasis HLB小柱凈化;另外,段科等[5]對(duì)肉中的阿托品類(lèi)藥物殘留量測(cè)定,乙腈提取后正己烷除油凈化。采用固相萃取柱凈化,步驟復(fù)雜且實(shí)驗(yàn)成本較高,而乙腈作為提取液,沒(méi)有兼容到強(qiáng)極性物質(zhì)的提取,對(duì)偏極性的蓄水類(lèi)藥物不能保證回收率。如何把極性物質(zhì)從水溶液中有效提取出來(lái),這始終是一個(gè)艱巨挑戰(zhàn)。隨著質(zhì)譜的發(fā)展和推廣,一次進(jìn)樣完成多類(lèi)藥物殘留分析已成為可能。藥物殘留正逐漸由液相色譜-三重四極桿質(zhì)譜(LC-MS/MS)目標(biāo)型檢測(cè)向高分辨質(zhì)譜(HRMS)非目標(biāo)型全掃描檢測(cè)轉(zhuǎn)變。與傳統(tǒng)的LC-MS/MS法相比較,高分辨質(zhì)譜儀可直接采集高準(zhǔn)確度質(zhì)量數(shù)(m/z200,分辨率70 000),大大降低了近似質(zhì)量數(shù)的干擾,通過(guò)自動(dòng)觸發(fā)二級(jí),避免了假陽(yáng)性結(jié)果的發(fā)生,特別適合高通量的篩查檢測(cè)。本文利用載體輔助液-液萃取技術(shù),通過(guò)一次前處理實(shí)現(xiàn)了9種常見(jiàn)注水類(lèi)藥物的提取凈化,同時(shí)結(jié)合四極桿靜電場(chǎng)軌道阱高分辨質(zhì)譜檢測(cè),為提高監(jiān)管工作效率,保證動(dòng)物源性食品安全提供了基礎(chǔ)。
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
Q-Exactive四極桿靜電場(chǎng)軌道阱高分辨質(zhì)譜儀(賽默飛世爾科技公司),配有H-ESI Ⅱ源。UltiMate 3000液相色譜系統(tǒng),配有自動(dòng)進(jìn)樣器。硅藻土柱型號(hào)為Chromabond XTR(1 000 mg)。
9種蓄水類(lèi)藥物標(biāo)準(zhǔn)品購(gòu)自Sigma和Dr.Ehrenstorfer公司,純度≥95%。二甲基亞砜(分析純)購(gòu)自南京化學(xué)試劑一廠。提取液的配制:稱(chēng)取4.3 g的H2C2O4,用500 mL水溶解,氨水調(diào)節(jié)pH至3.0。色譜純甲酸購(gòu)自美國(guó)Sigma-Aldrich公司。其他試劑均為色譜純,購(gòu)自德國(guó)Merck公司。實(shí)驗(yàn)用水為Milli-Q超純水(18.2 ΩM·cm)。
1.2 提取凈化流程
基質(zhì)樣品來(lái)自國(guó)家豬肉殘留監(jiān)控抽樣和進(jìn)出口送檢企業(yè)。稱(chēng)取5.00 g樣品,加入10 mL提取液,振蕩、各超聲5 min,4 500 r/min下離心5 min;取水相上大孔硅藻土柱后,平衡5 min,用10 mL乙腈洗脫2次,下接50 mL離心管,并加入0.5 mL二甲基亞砜,乙腈定容至20 mL;取出10 mL,氮吹后用純水定容至1.0 mL,過(guò)0.22 μm 濾膜,待分析。
1.3 色譜及質(zhì)譜條件
色譜柱:Hypersil Gold C18柱(100 mm×2.1 mm,1.9 μm);流動(dòng)相A:0.1%甲酸水溶液;流動(dòng)相B:乙腈。梯度洗脫:0~3 min,B相保持5%;3~6 min,B相5%~90%;6~8 min,B相保持90%;8~8.1 min,B相90%~5%。流速0.3 mL/min;柱溫40 ℃;進(jìn)樣量10 μL。
質(zhì)譜條件:質(zhì)譜在正/負(fù)離子轉(zhuǎn)換模式下進(jìn)行全掃描,質(zhì)量范圍:m/z100~500,分辨率70 000,自動(dòng)增益控制(AGC)目標(biāo)值5.0×105;4-羥基-3-甲氧基-扁桃酸和3,4-二羥基扁桃酸采用負(fù)離子模式2 700 V,其余成分則采用正離子模式3 800 V;離子傳輸管溫度為300 ℃;鞘氣流速(N2)35 L/h;輔助流速(N2)10 L/h;氣化室溫度350 ℃。在樣品運(yùn)行前對(duì)儀器分別進(jìn)行正、負(fù)離子校正;二級(jí)采用自動(dòng)觸發(fā)模式,分辨率35 000,AGC目標(biāo)值2.0×105,碰撞能量范圍25%~40%,保留時(shí)間采集范圍:根據(jù)一級(jí)色譜圖中各個(gè)目標(biāo)物的保留時(shí)間值±1.0 min。
1.4 定性、定量分析
定性分析時(shí)精確質(zhì)量誤差低于5.0×10-6,同時(shí)比對(duì)保留時(shí)間、同位素分布、主要二級(jí)碎片和二級(jí)質(zhì)譜圖相似度,綜合判斷,避免假陽(yáng)性結(jié)果出現(xiàn)。
空白樣品按照“1.2”處理,在得到的基質(zhì)溶液中加入上述9種目標(biāo)物混合標(biāo)準(zhǔn)溶液,配制基質(zhì)加標(biāo)溶液并進(jìn)行測(cè)定,以濃度為橫坐標(biāo),儀器響應(yīng)值(響應(yīng)值=標(biāo)準(zhǔn)品峰面積/標(biāo)準(zhǔn)品質(zhì)量)為縱坐標(biāo)做標(biāo)準(zhǔn)曲線,作為樣品中待測(cè)物濃度定量的依據(jù)。其中,高濃度加標(biāo)樣品應(yīng)稀釋至標(biāo)準(zhǔn)曲線范圍內(nèi)進(jìn)行定量分析。
2 結(jié)果與討論
2.1 提取液的選擇
考慮到豬肉基質(zhì)的高脂肪屬性,且9種待測(cè)物均為堿性化合物,水溶性強(qiáng),方法采用酸化的水溶液進(jìn)行提取,在保證9種蓄水類(lèi)化合物高提取率的同時(shí),又可以防止脂肪被過(guò)多的提取出來(lái),降低脂肪的干擾。H2C2O4含有的兩個(gè)羧基基團(tuán),較其他可揮發(fā)性甲酸、乙酸,具有更強(qiáng)的穩(wěn)定性和酸性,此外H2C2O4作為金屬螯合劑,可大大降低金屬離子的干擾。
2.2 提取劑的添加量對(duì)柱填料的影響
提取液的添加量要充分考慮柱子填料的含量,本文選擇硅藻土柱Chromabond XTR(1 000 mg)。實(shí)驗(yàn)中分別比較了上樣量為5 mL、8 mL、10 mL、12 mL、15 mL時(shí)柱子的負(fù)載情況,在上樣量達(dá)到12 mL和15 mL時(shí),柱子已達(dá)到飽和并滲出,水溶液的滲出會(huì)導(dǎo)致氮吹濃縮過(guò)程大大延長(zhǎng)。另外,當(dāng)上樣量為5 mL和8 mL時(shí),柱子的填料未充分浸入水溶液,大大降低了柱子的利用率,同時(shí)對(duì)于9種待測(cè)藥物本就很低的檢出限量來(lái)說(shuō),也增加了儀器檢出的難度。因此,本研究最終將上樣體積確定為10 mL。

圖1 乙腈洗脫體積對(duì)9種注水類(lèi)藥物回收率的影響Fig.1 Effect of elution volume of acetonitrile on recovery of 9 waterflooding drugs
2.3 洗脫體積的優(yōu)化
水相提取液上樣后,以純乙腈作為洗脫液,通過(guò)乙腈的逐級(jí)滲入,注水類(lèi)藥物按照極性強(qiáng)度依次從飽和的硅藻土柱上洗脫下來(lái)。實(shí)驗(yàn)中分別考察乙腈作為洗脫溶劑的添加量為10 mL、10 mL×2和10 mL×3對(duì)待測(cè)物回收率的影響。由圖1可見(jiàn),當(dāng)乙腈的體積為10 mL×2和10 mL×3時(shí),提取率趨于穩(wěn)定,9種待測(cè)注水類(lèi)藥物在豬肉中的回收率均保持在80%以上。從節(jié)約氮吹濃縮的時(shí)間和成本的角度出發(fā),確定純乙腈作為洗脫液的洗脫體積為10 mL×2。
2.4 儀器條件優(yōu)化
通過(guò)注射泵連續(xù)進(jìn)樣,對(duì)上述蓄水類(lèi)藥物的單標(biāo)準(zhǔn)溶液進(jìn)行分析,獲得每種化合物的最佳電離方式和分子離子峰的存在形式。其中4-羥基-3-甲氧基-扁桃酸和3,4-二羥基扁桃酸采用ESI-模式,其余化合物均采用ESI+模式。
流動(dòng)相中加入0.1%甲酸能增加正離子模式檢測(cè)下物質(zhì)的電離效率,促進(jìn)[M+H]+離子生成,同時(shí)進(jìn)一步考察NH4Ac添加量對(duì)待測(cè)化合物的影響,NH4Ac從2 mmol/L到10 mmol/L,結(jié)果表明9種蓄水類(lèi)藥物在0.1%甲酸溶液中均獲得了最佳的色譜峰形、分離效果和質(zhì)譜信號(hào)響應(yīng)。從減小儀器污染的角度出發(fā),本文最終采用含有0.1%甲酸水溶液作為流動(dòng)相A,全程采用梯度洗脫,實(shí)現(xiàn)了9種待測(cè)物的有效分離。9種注水類(lèi)藥物的提取離子色譜圖見(jiàn)圖2。
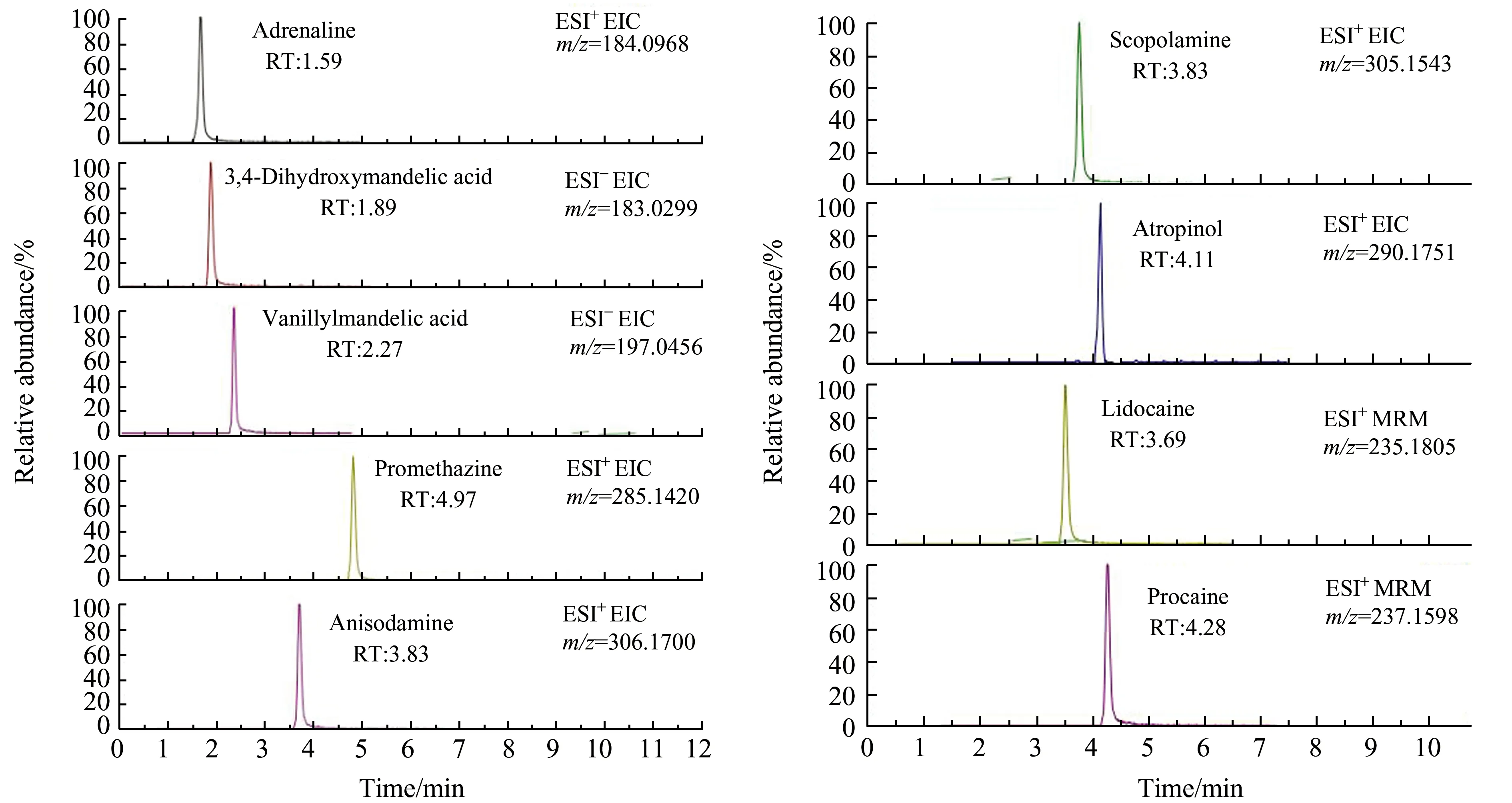
圖2 9種蓄水藥物的提取離子色譜圖Fig.2 Extraction ion chromatograms of 9 waterflooding drugs
2.5 加標(biāo)回收結(jié)果分析
結(jié)果表明,腎上腺素及其代謝物4-羥基-3-甲氧基-扁桃酸在5.0~50.0 ng/mL濃度范圍內(nèi),其他藥物(異丙嗪,阿托品、山莨菪堿、東莨菪堿、普魯卡因和利多卡因)在0.5~5.0 ng/mL濃度范圍內(nèi),各自呈現(xiàn)良好的線性關(guān)系,相關(guān)系數(shù)r2均大于0.990,滿足定量要求。9種待測(cè)物的回收率在80.0%~95.2%之間,相對(duì)標(biāo)準(zhǔn)偏差低于10%。用基質(zhì)提取液添加低水平的標(biāo)準(zhǔn)溶液(腎上腺素及代謝物為5.0 μg/kg,其他待測(cè)物為0.5 μg/kg),獲得每種待測(cè)物對(duì)應(yīng)信噪比(S/N)≥10時(shí)的含量,以此為該化合物的定量限(LOQ)(表1)。結(jié)果表明,該方法靈敏度高,操作簡(jiǎn)便,適用于豬肉中蓄水類(lèi)藥物殘留的快速篩查。
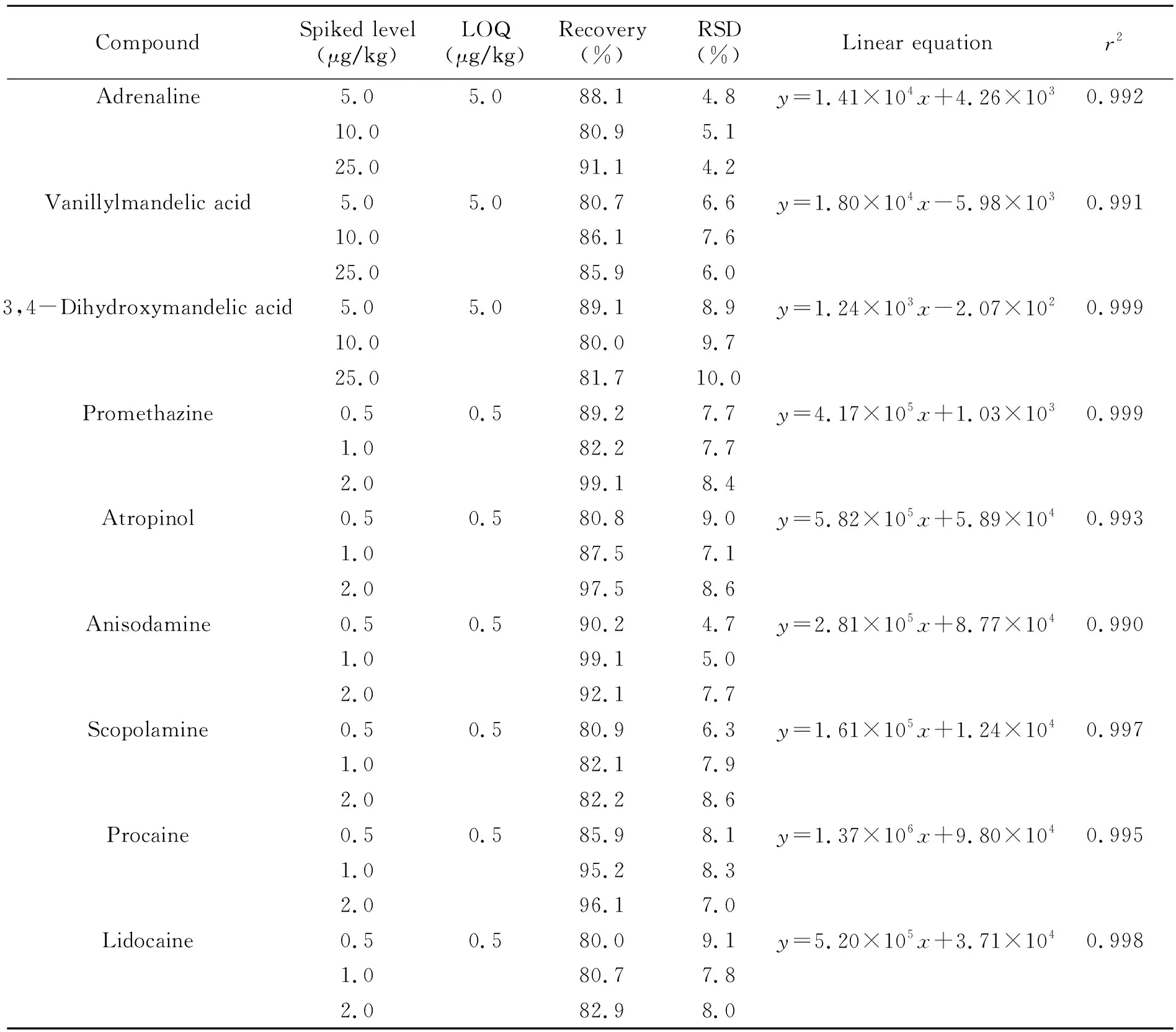
表1 豬肉中9種注水類(lèi)藥物的定量限(LOQs)、線性方程及不同添加濃度的平均回收率(n=6)
3 結(jié)論
本研究建立了豬肉中常見(jiàn)的9種蓄水類(lèi)藥物:腎上腺素及其代謝物4-羥基-3-甲氧基-扁桃酸和3,4-二羥基扁桃酸、異丙嗪、阿托品、山莨菪堿、東莨菪堿、普魯卡因和利多卡因的快速篩查方法。待測(cè)物經(jīng)水溶液提取后,利用硅藻土柱完成水溶性藥物的同時(shí)提取、凈化、濃縮過(guò)程。同時(shí)結(jié)合四極桿靜電場(chǎng)軌道阱高分辨質(zhì)譜,實(shí)現(xiàn)了上述藥物殘留的“一步式”多殘留篩查。該方法操作簡(jiǎn)單,重現(xiàn)性好,定量限低,完全滿足國(guó)標(biāo)中規(guī)定的豬肉中9種蓄水類(lèi)藥物的測(cè)定需求。

- 分析科學(xué)學(xué)報(bào)的其它文章
- 《分析科學(xué)學(xué)報(bào)》征稿簡(jiǎn)則
- 《分析科學(xué)學(xué)報(bào)》編輯委員會(huì)
- 稀土元素?fù)诫s上轉(zhuǎn)換納米材料在衛(wèi)生分析領(lǐng)域中的應(yīng)用
- 電感耦合等離子體質(zhì)譜法測(cè)定金屬單元素溶液標(biāo)準(zhǔn)物質(zhì)中雜質(zhì)
- 高效液相色譜-串聯(lián)質(zhì)譜法同時(shí)測(cè)定雞肉和雞蛋中10種硝基咪唑類(lèi)藥物及代謝物
- QuEChERS-氣相色譜-串聯(lián)質(zhì)譜法同時(shí)測(cè)定銀耳中17種殺蟲(chóng)劑殘留