離子色譜法測定口服電解質顆粒中陰、陽離子的含量
2021-05-08 05:40:42張建茹昝艷楠丁逸梅
藥學與臨床研究 2021年2期
張建茹,昝艷楠,2,丁逸梅,2*
1 南京工業大學 藥學院,南京 211800;2 江蘇省藥物研究所,南京 211800
人體的電解質大部分存在于細胞內外,保持著人體的平衡,電解質參與體內許多重要的功能和代謝活動,對維持正常生命活動起著重要的作用。本文中的口服電解質顆粒原研廠家為日本味之素株式會社,目前國內沒有上市。本品為氯化鈉、氯化鉀、無水磷酸二氫鈉、檸檬酸鈉水合物、碳酸鎂、水合檸檬酸、香料、乙基香草醛、香蘭素、丙二醇等組成的顆粒,用于補充身體所需的離子。
氯離子、磷酸根離子及檸檬酸根離子的常規含量測定方法有電位滴定法[1]、高效液相色譜法[2]、氣相色譜法[3]、離子色譜[4,5]等。電位滴定法每次只能測定一種離子,消耗時間長且測定要求較高(需要相應的專用電極);而檸檬酸根在高效液相中保留較弱,檢測器靈敏度低;而用氣相色譜法測定需衍生化等過程,操作復雜。使用離子色譜法可同時測定多種離子,且制備樣品簡單。有報道采用離子色譜法進行檸檬酸測定[6],由于檸檬酸是弱有機酸,在離子色譜柱上保留較強,常用梯度洗脫方式與其他常見陰離子進行分離,但分析時間較長,對設備配置要求高。
目前鈉、鉀、鎂的測定方法主要有原子吸收法[3,4]、電感耦合等離子體發射光譜法(ICP-MS)[5]、滴定法[6]、電極法[7]、離子色譜法等。原子吸收法測定金屬時要進行預處理,加入消電離劑、釋放劑等,且每次只能分析一種元素。滴定法和電位法操作較為繁瑣,靈敏度不高,會造成二次污染,且當多種元素同時存在時,滴定法無法同時區分。ICP-MS 雖然靈敏度高,分析速度快,但其使用和維護成本比較高,儀器不夠普及,并且不能直接進樣進行分析[8]。
本文建立了同時快速測定氯離子、磷酸根離子、檸檬酸根離子、鈉離子、鉀離子、鎂離子含量的等度洗脫離子色譜法,具有操作簡便,直接進樣,靈敏度高等優點,是目前最常用的陰陽離子分析方法。
1 儀器與藥品、試劑
1.1 儀器
930 Compact IC Flex 離子色譜儀(瑞士萬通,配自動淋洗液發生器,CO2抑制器MCS,MagIC Net 3.2 色譜工作站);陰離子色譜柱:Metrosep A SUPP 5 column(50 mm×4 mm),陰離子保護柱:Metrosep A SUPP 5 Guard/4.0;陽離子色譜柱:Metrosep C4column(250 mm×4 mm),陽離子保護柱:Metrosep C4Guard/4.0;On-GuardH 柱及C18預處理小柱(美國戴安公司);萬分之一天平(Sartorius,BT25S);十萬分之一天平(Sartorius,BT25S)。
1.2 藥品與試劑
氯化鉀對照品(純度99.8%,批號:20190414)、六水合氯化鎂(純度99.8%,批號:20180804),均為國藥集團化學試劑有限公司;氯化鈉對照品(純度99.8%,批號:20180619)、一水合檸檬酸(純度99.8%,批號:20180313)、氯化鈉(純度99.8%,批號:20180619),均為南京化學試劑公司;磷酸二氫鈉(純度99.8%,批號:20180804,永華化學科技公司);口服電解質:商品名Solita-T?granules No.3(批號:4F371、4F373、4F375、4F377、4F379,日本味之素制藥);硝酸(純度100.0%)、碳酸鈉(純度100.0%)分析純,市售;超純水和二次去離子水(用超聲波脫氣)。
2 方法和結果
2.1 陰離子(氯、磷酸根、檸檬酸根離子)測定
2.1.1 供試品溶液的制備取本品5 包稱定,研細,精密稱取細粉適量,置50 mL 量瓶中,加水適量超聲使電解質顆粒溶解,用水稀釋至刻度,搖勻,作為儲備液(約每毫升含氯離子1065 μg、磷酸根475 μg、檸檬酸根2160 μg)。取儲備液2.0 mL 置50 mL 量瓶中,用水稀釋至刻度,作為供試品溶液。
2.1.2 對照品的制備精密稱取一水合檸檬酸、氯化鈉、磷酸二氫鈉對照品適量,加水適量使溶解并稀釋成每毫升約含氯離子1065 μg,磷酸根475 μg和檸檬酸根2160 μg 的混合對照品儲備溶液,取該儲備液2.0 mL,置50 mL 量瓶中,加水稀釋至刻度,作為混合對照品溶液。
2.1.3 色譜條件與系統適用性試驗取一水合檸檬酸、氯化鈉、磷酸二氫鈉對照品適量,加水適量使溶解并稀釋成每毫升約含氯離子42.60 μg、磷酸根19 μg 和檸檬酸根86.4 μg 的溶液作為系統適用性溶液,采用Metrosep A Supp-5 column(150 mm×4 mm)色譜柱;流動相:碳酸鈉溶液(12.8 mmol·L-1)、等度洗脫;柱溫:30 ℃;流速:0.7 mL·min-1;進樣量:20 μL;電導檢測器。在上述條件下,各峰之間的分離度良好(Rs>1.5),出峰順序依次為氯離子、磷酸根、檸檬酸根。色譜圖見圖1。
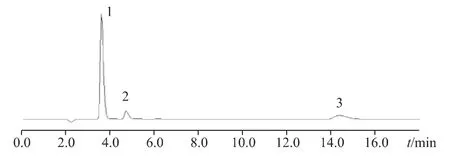
圖1 系統適用性色譜圖
2.1.4 專屬性取處方中不含3 種陰離子的其他成分,稀釋成與供試品、對照品相同倍數的空白溶液,與混合對照品、供試品一并分析。結果顯示,本方法測定處方中3 種陰離子的專屬性良好,不受溶液中其他成分(陽離子、香料、乙基香草醛、香蘭素、丙二醇等)的干擾,且空白輔料和溶劑中沒有干擾峰,各陰離子之間分離度良好。色譜圖見圖2。
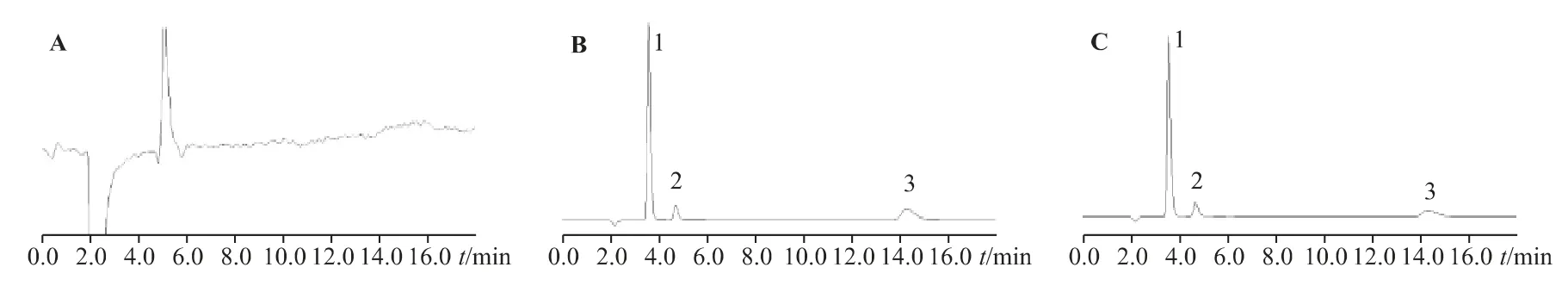
圖2 空白溶液離子(A)、標準溶液陰離子(B)、供試品溶液陰離子(C)色譜圖
2.1.5 線性關系考察精密量取對照品儲備液1、1.4、2.0、2.6、3 mL 分別置于50 mL 量瓶中,用水稀釋至刻度,得相當于氯離子濃度分別為21.30、29.82、42.60、55.38、63.90 μg·mL-1,磷酸根濃度分別為9.500、13.30、19.00、24.70、28.50 μg·mL-1,檸酸根濃度分別為43.20、60.48、86.40、112.3、129.6 μg·mL-1的系列對照品溶液。按“2.1.3”項下色譜條件進樣,以樣品濃度(X,μg·mL-1)為橫坐標,峰面積(Y)為縱坐標,擬合回歸方程見表1。
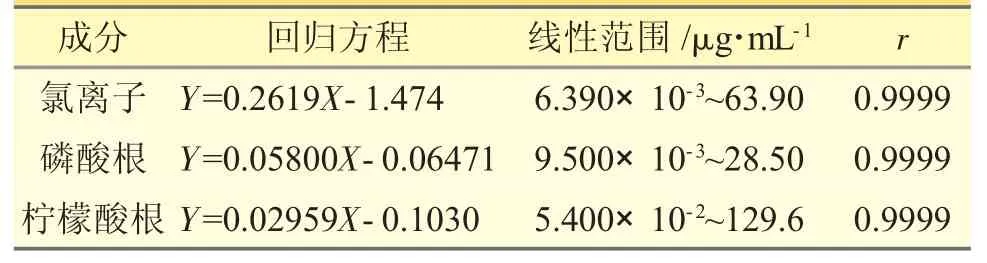
表1 陰離子含量測定法的線性范圍
2.1.6 檢測限和定量限取“2.1.2”項下對照品適量,等倍逐步稀釋,按“2.1.3”項下色譜條件連續進樣6 次,記錄峰面積。當信噪比為10∶1 時,得定量限;當信噪比為3∶1 時,得檢測限。結果LOD 和LOQ氯離子分別為2.130 ng·mL-1和6.390 ng·mL-1;磷酸根分別為3.160 ng·mL-1和9.500ng·mL-1;檸檬酸根分別為54ng·mL-1和162 ng·mL-1。
2.1.7 進樣精密度試驗取“2.1.2”項下對照品溶液20 μL,按“2.1.3”項下色譜條件進樣測定,記錄色譜圖。結果氯離子、磷酸根、檸檬酸根的RSD 分別為0.14%、0.30%、0.52%(n=6),表明儀器精密度良好。
2.1.8 穩定性試驗取“2.1.1”項下供試品溶液,在室溫下放置0、2、4、6、8、12、24 h,分別進樣測定,記錄色譜圖。結果氯離子、磷酸根、檸檬酸的峰面積RSD 分別為0.23%、1.6%、0.37%,表明供試品溶液在室溫24h 內穩定。取“2.1.2”項下對照品溶液,以下操作同上。結果氯離子、磷酸根、檸檬酸的峰面積RSD 分別為0.55%、1.8%、0.29%,表明對照品溶液在室溫下24 h 內穩定。
2.1.9 重復性試驗取“2.1.1”項下供試品6 份,再按“2.1.3”項下色譜條件連續進樣,記錄色譜圖。結果,6 份供試品溶液中氯離子、磷酸根、檸檬酸的平均含量分別為42.11、18.48、84.98 μg·mL-1,它們的RSD分別為1.3%、0.8%、1.6%,表明本方法重現性良好。
2.1.10 加樣回收率試驗精密稱定本品0.04 g,置50 mL 量瓶中,共9 份,精密加入氯離子、磷酸根、檸檬酸根質量濃度分別為1065、475、2160 μg·mL-1的混合對照品溶液7.0、8.0、10.0 mL 各3 份,按照“2.2.1”項下方法制備供試品溶液,得低、中、高(70%、100%、130%)3 個濃度,注入離子色譜儀進行測定,計算回收率,本方法的準確度良好。見表2。
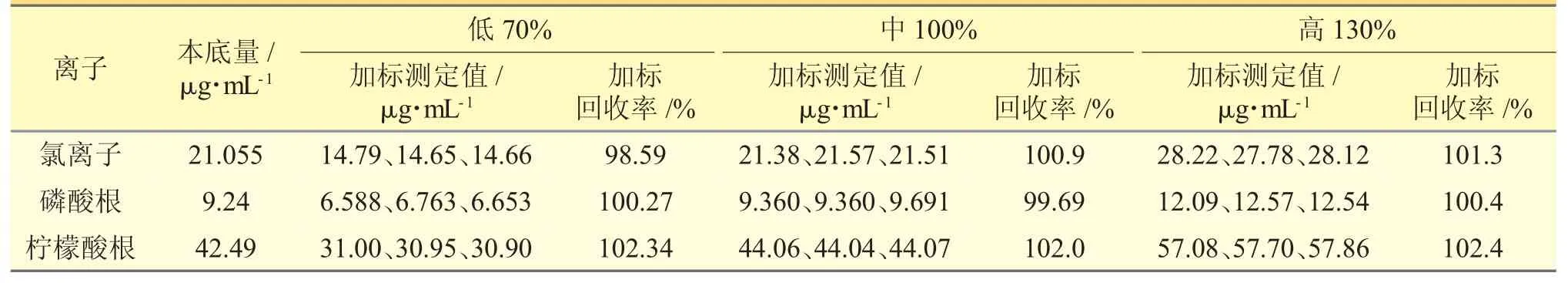
表2 加樣回收率試驗結果
2.2 陽離子(鈉離子、鉀離子、鎂離子)測定
2.2.1 供試品溶液的制備取本品5 包,稱定,研細,精密稱取細粉適量,置100 mL 量瓶中,加水適量超聲振蕩使電解質顆粒溶解,用水稀釋至刻度,搖勻,作為儲備液(約每毫升含鈉離子805μg、鉀離子780 μg、鎂離子36 μg)。取儲備液2.0 mL 置50 mL量瓶中,用水稀釋至刻度,作為供試品溶液。
2.2.2 對照品的制備精密稱取氯化鈉、氯化鉀、六水合氯化鎂對照品,置于100 mL 量瓶,加水適量溶解并稀釋成每毫升約含鈉離子805 μg、鉀離子780 μg 和鎂離子36 μg 的混合對照品儲備液,取儲備液2.0 mL,置50 mL 量瓶中,加水稀釋至刻度,作為混合對照品溶液。
2.2.3 色譜條件與系統適用性試驗精密稱取氯化鈉、氯化鉀、六水合氯化鎂對照品置于100 mL 量瓶,加水適量使溶解并稀釋成每毫升約含鈉離子32.20 μg、鉀離子31.20 μg 和鎂離子1.44 μg 的混合對照品儲備液,采用Metrosep A Supp-5 column(250mm×4mm)色譜柱;流動相為硝酸溶液(12.8mmol·L-1),等度洗脫;柱溫:30 ℃;流速:0.7 mL·min-1;進樣量:20 μL;電導檢測器。在上述條件下,各峰之間的分離度良好(Rs>1.5),出峰順序依次為鈉離子、鉀離子、鎂離子,且分離度良好。色譜圖見圖3。
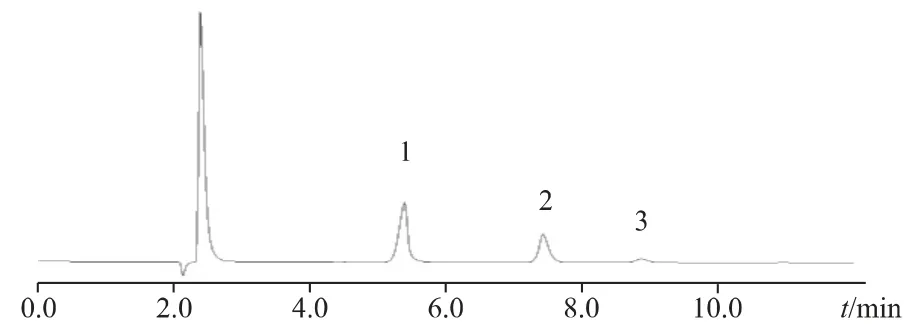
圖3 系統適用性色譜圖
2.2.4 專屬性取處方中不含3 種陽離子的其他成分,稀釋成與供試品、對照品相同倍數的空白溶液,與混合對照品、供試品一并分析。結果顯示,本方法測定處方中3 種陽離子的專屬性良好,不受溶液中其他成分(陰離子、香料、乙基香草醛、香蘭素、丙二醇等輔料)的干擾,因此空白溶液中沒有干擾峰,各陽離子之間分離度良好。色譜圖見圖4。
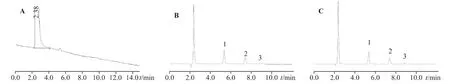
圖4 空白溶液離子(A)、標準陽離子溶液(B)、供試品溶液陽離子(C)色譜圖
2.2.5 線性關系考察精密量取“2.2.2”項下對照品儲備液1、1.4、2.0、2.6、3 mL 分別置于50 mL 量瓶中,用水稀釋至刻度,得相當于鈉離子濃度分別為16.07、22.50、32.15、41.79、48.22 μg·mL-1,鉀離子濃度分別為15.69、21.97、31.39、40.80、47.08 μg·mL-1,鎂離子濃度分別為0.7290、1.021、1.458、1.896、2.187 μg·mL-1的系列對照品溶液。按“2.2.3”項下色譜條件進樣,以樣品濃度(X,μg·mL-1)為橫坐標,峰面積(Y)為縱坐標,擬合回歸方程見表3。
2.2.6 檢測限和定量限取“2.2.2”項下對照品溶液適量,等倍逐步稀釋,按“2.2.3”項下色譜條件連續進樣測定6 次,記錄峰面積。當信噪比為10∶1 時,得定量限;當信噪比為3∶1 時,得檢測限。結果LOD 和LOQ 鈉離子分別為0.0321μg·mL-1和0.107μg·mL-1;鉀離子分別為0.03138 μg·mL-1和9.500 μg·mL-1;鎂離子分別為0.001458 μg·mL-1和0.00486 μg·mL-1。
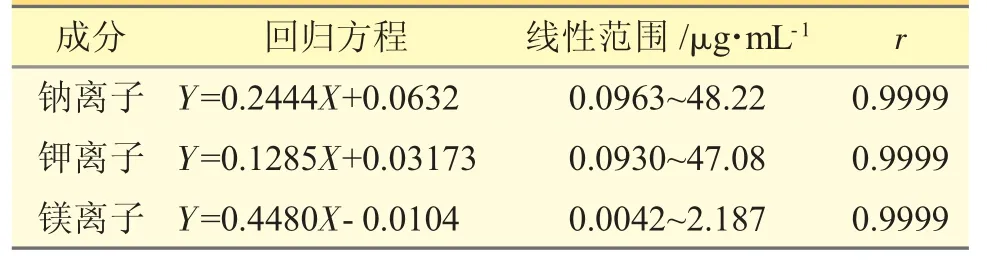
表3 陽離子含量測定法的線性范圍
2.2.7 精密度試驗取“2.2.2”項下對照品溶液20μL,按“2.2.3”項下色譜條件連續進樣測定,記錄色譜圖。結果RSD 鈉離子為0.56%、鉀離子為0.65%、鎂離子為0.97%(n=6),表明儀器精密度良好。
2.2.8 穩定性試驗取“2.2.1”項下供試品溶液,在室溫下放置0、2、4、6、8、12、24 h,分別進樣測定,記錄色譜圖。結果鈉離子、鉀離子、鎂離子的峰面積RSD分別為0.32%、0.31%、1.27%,表明供試品溶液在室溫24 h 內穩定;取“2.2.2”項下對照品溶液,以下同上操作。結果鈉、鉀、鎂離子的峰面積RSD 分別為0.39%、0.32%、1.66%,表明對照品在室溫24h 內穩定。
2.2.9 重復性試驗取“2.2.1”項下供試品6 份,按“2.2.3”項下色譜條件進樣測定,記錄色譜圖,以外標法計算鈉、鉀、鎂離子的含量,其含量的RSD 分別為0.2%、0.25%、0.08%,表明本方法重現性良好。
2.2.10 加樣回收率試驗精密稱定本品0.04 g,置50 mL 量瓶中,共9 份,精密加入鈉、鉀、鎂離子質量濃度分別為805、780、36 μg·mL-1的混合對照品溶液0.4、1.0、1.6 mL,各3 份,按照“2.2.1”項下方法制備供試品溶液,得低、中、高(70%、100%、130%)3 個濃度,注入離子色譜儀進行測定,計算回收率,本方法的準確度良好。見表4。
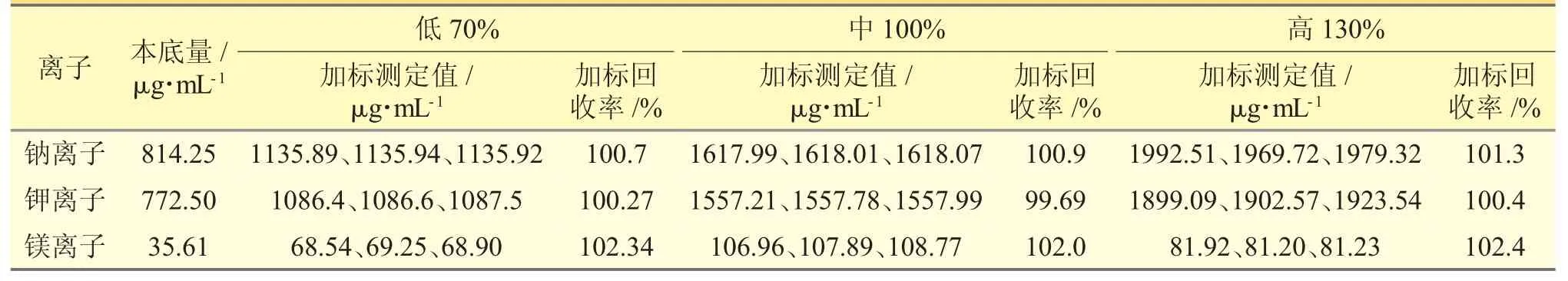
表4 加樣回收率試驗結果
2.3 樣品測定
對5 批樣品照上述方法測定,按外標法計算各離子的含量,結果見表5。
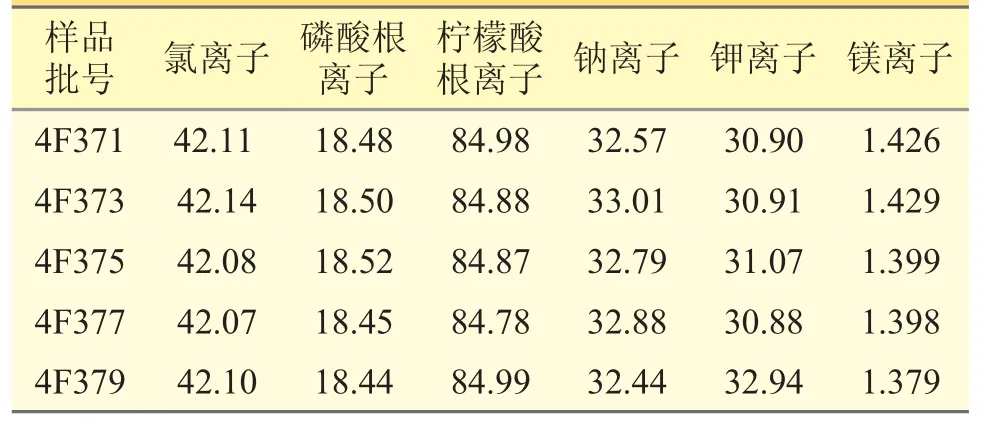
表5 樣品含量測定結果(μg·mL-1)
3 討論
3.1 淋洗液條件的選擇
分離效果一方面取決于待測離子的濃度,另一方面取決于淋洗液的濃度,即淋洗液的濃度影響著分離時間和分離度。對于陰離子分析,本實驗比較了7 mmol·L-1Na2CO3+3 mmol·L-1NaHCO3+5%丙酮(圖5A)、7 mmol·L-1Na2CO3+3 mmol·L-1NaHCO3+25%丙酮(圖5B)及不用丙酮,改為12.8 mmol·L-1Na2CO33 種淋洗液對分離效果的影響。
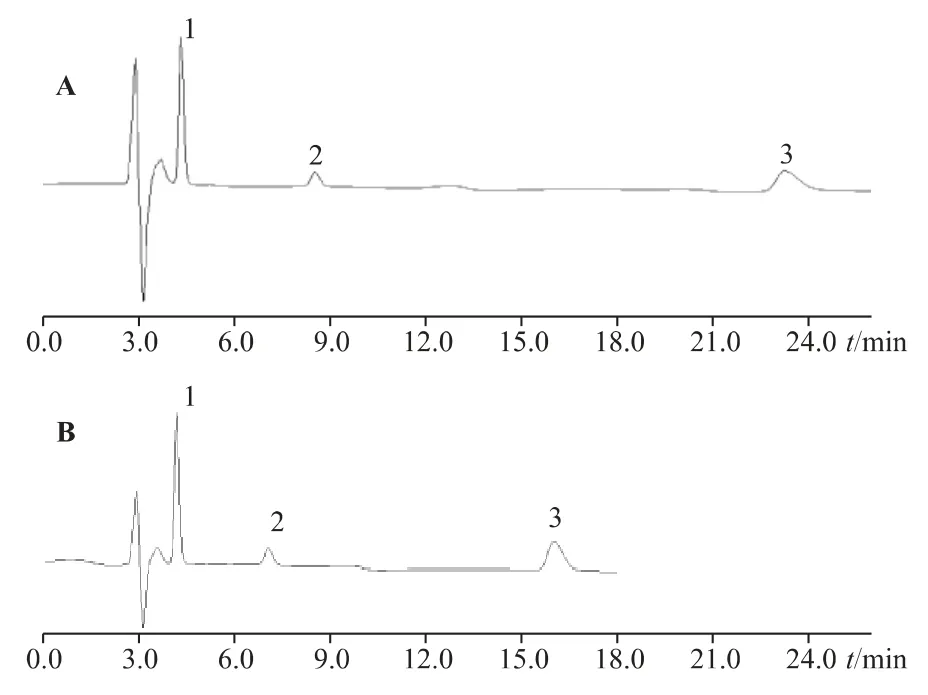
圖5 不同淋洗條件下的色譜圖
圖5A 與圖6B 相比,隨著丙酮濃度增加,檸檬酸保留時間縮短,峰形變得尖銳。但因加入高濃度的丙酮后,基線漂移嚴重,且隨著進樣次數增加,保留時間越來越延后,還會出現抑制器飽和的現象,故不加入丙酮,逐漸改變Na2CO3的濃度,最后確定12.8 mmol·L-1Na2CO3作為淋洗液進行陰離子分子最為合適,經濟適用且環保。
在使用離子色譜測定陽離子時,大部分文獻所采用的的流動相是20 mmol·L-1甲基磺酸,本實驗采用5 mmol·L-1硝酸便可使各個離子得到很好的分離。實驗過程中考察了1、5、10、15 mmol·L-1硝酸對鈉、鉀、鎂的分離效果。結果顯示,5 mmol·L-1硝酸即可實現鈉鉀鎂的有效分離,且分離度>1.5。見表6。
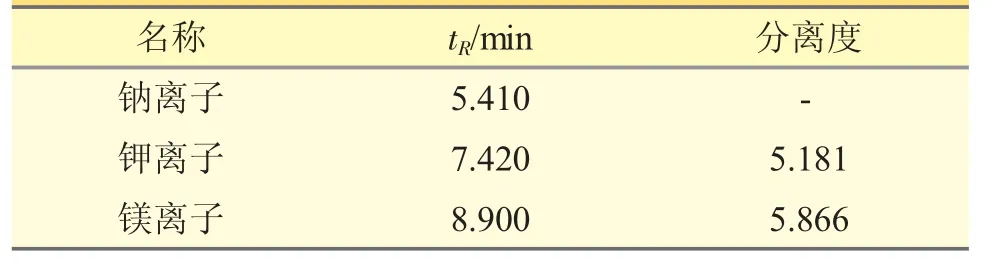
表6 標準淋洗液流速下各陽離子色譜峰分離度
3.2 抑制器轉換
過高濃度的淋洗液會使抑制器容量飽和,實驗使用了在采集時間內對抑制器進行命令,使其8 分鐘切換一次,避免抑制器飽和致使基線抬高,背景增大,且切換時間在磷酸根與檸檬酸根出峰時間之間,避免了對離子出峰的干擾。
3.3 檸檬酸根離子與等度洗脫
檸檬酸是弱有機酸,在離子色譜柱上保留較強,常用梯度洗脫方式與其他常見陰離子進行分離,但分析時間較長,對設備配置要求高。本實驗采用簡單的等度洗脫進行檸檬酸根測定,其離子能快速出峰,且峰型平穩,有效避免了梯度洗脫使基線不平穩,對儀器的配置要求高(通常需要配置四元泵或者二元泵)的麻煩。
