氫氧直接合成過氧化氫用催化劑的研究進展
2021-04-20 10:31:54梁海瑞王涖劉國柱
化工進展 2021年4期
關(guān)鍵詞:催化劑
梁海瑞,王涖,劉國柱
(1 中海石油氣電集團有限責(zé)任公司,北京100028;2 天津大學(xué)化工學(xué)院綠色合成與轉(zhuǎn)化教育部重點實驗室,天津300072)
過氧化氫(H2O2)為無色透明液體,具有較強的氧化能力,氧化過程中,自身生成水,無其他副產(chǎn)物,是一種綠色無污染的強氧化劑[1]。由于工業(yè)對過氧化氫有較強的需求,過氧化氫被廣泛應(yīng)用于化學(xué)品的合成、印染造紙、污水處理等各個領(lǐng)域。過氧化氫的制備方法主要包括電化學(xué)法、蒽醌法、異丙醇法和氫氧直接合成法等[2]。目前,由于蒽醌法安全高效等優(yōu)勢,工業(yè)上95%過氧化氫生產(chǎn)方法為蒽醌法,但蒽醌法制備過氧化氫也存在著能耗、環(huán)境等問題。直接合成過氧化氫是指氫氣和氧氣直接反應(yīng)產(chǎn)生過氧化氫,該反應(yīng)一般由貴金屬催化劑催化,過程的唯一副產(chǎn)物是水。相對于現(xiàn)有的復(fù)雜蒽醌工藝,該過程只需一步反應(yīng),避免了有害的反應(yīng)條件、化學(xué)品和副產(chǎn)物,是一種清潔的工藝過程,因而在商業(yè)上有良好的前景。本文從催化直接合成過氧化氫反應(yīng)途徑、催化用金屬、催化用載體等方面,主要總結(jié)了近五年來直接合成過氧化氫用催化劑的研究進展。
1 直接合成過氧化氫反應(yīng)途徑
直接催化合成過氧化氫的過程存在4種化學(xué)反應(yīng)(見表1)。對比各反應(yīng)的吉布斯自由能可看出,H2和O2直接生成H2O的反應(yīng)、H2O2加氫反應(yīng)、H2O2分解反應(yīng)與生成H2O2的反應(yīng)存在明顯的競爭。

表1 直接合成過氧化氫過程中存在的反應(yīng)
目前,貴金屬催化劑是氫氧直接合成過氧化氫反應(yīng)中使用最多的催化劑。針對貴金屬的催化機理研究中,普遍認為H2O2的合成過程存在著以O(shè)OH為中間體的兩步氫化過程。Lunsford等[4]利用同位素分析直接合成H2O2過程,通過提供18O2/16O2=1∶1,分析發(fā)現(xiàn)反應(yīng)產(chǎn)物中不存在H18O16OH,說明在反應(yīng)過程中O2并不存在解離,直接合成過氧化氫過程為氧分子與氫原子非解離加氫生成H2O2。目前普遍認為的反應(yīng)路徑為:首先,H2和O2分別在金屬顆粒表面吸附;其次,H2在金屬表面解離,吸附的O2得到H2解離釋放的電子生成中間產(chǎn)物(OOH*,*為反應(yīng)活性位點);最后,生成的中間產(chǎn)物與解離后的H結(jié)合并從金屬表面脫附,得到H2O2(見圖1)。
2 直接合成過氧化氫催化劑用金屬
貴金屬催化直接合成過氧化氫的活性金屬一般有Pd、Au、Pt 等貴金屬以及它們的合金。H2O2合成過程中的活性和選擇性與各反應(yīng)物及反應(yīng)產(chǎn)物的吸附/脫附能和生成/解離活化能相關(guān)。Ford 等[6]基于周期性密度泛函理論(DFT-GGA)計算,采用周期性平板模型(Slab)計算得到若干不同金屬的(111)晶面的解離活化能如圖2 所示。Pd 和Pt 表面上OOH 中間體和H2O2的解離活化能與H2O2的生成活化能和脫附能相比更低,說明Pt(111)晶面上容易發(fā)生O—O鍵的斷裂,使得反應(yīng)選擇性低,而Au表面上H2O2的生成活化能和脫附能比OOH 中間體和H2O2的解離活化能要低,使得Au的選擇性較高,然而由于反應(yīng)物H2和O2在Au 表面的吸附能很高,使得Au反應(yīng)活性很低。
2.1 單金屬Pd催化直接合成過氧化氫
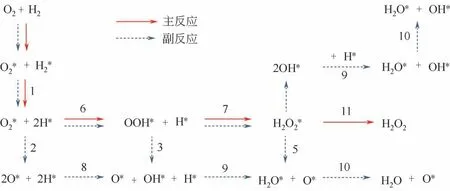
圖1 氫氧直接合成過氧化氫反應(yīng)途徑[5](*為反應(yīng)活性位點)
目前,氫氧直接合成過氧化氫的研究主流為Pd 基催化劑,針對Pd 金屬活性位的研究眾多。Tian等[7]通過DFT計算分析了Pd金屬不同晶面對于反應(yīng)能壘的影響,發(fā)現(xiàn)Pd(111)晶面與其他晶面如Pd(100)、Pd(110)等相比,Pd(111)晶面的配位飽和度更高,Pd(111)表面原子間d 電子軌道重疊較高,靠近費米能級(Fermi level)的電子態(tài)密度較小,從而更難反饋給O22π 反鍵軌道(2π*)電子,使得Pd 表面OOH 中間體和H2O2的解離活化能較高,即O—O鍵較難在Pd(111)表面解離,Pd(111)晶面具有最高的反應(yīng)選擇性;此外,在Pd 晶面的臺階位如Pd(211)表面的(100)臺階位,O2和H2O2的解離活化能均很低,且H2和O2吸附能很低,使得Pd晶面的臺階位選擇性很低。這一結(jié)論表明制備高選擇性催化劑需要抑制Pd顆粒臺階位(邊緣和角落位置)對于O2和H2O2的解離活化。
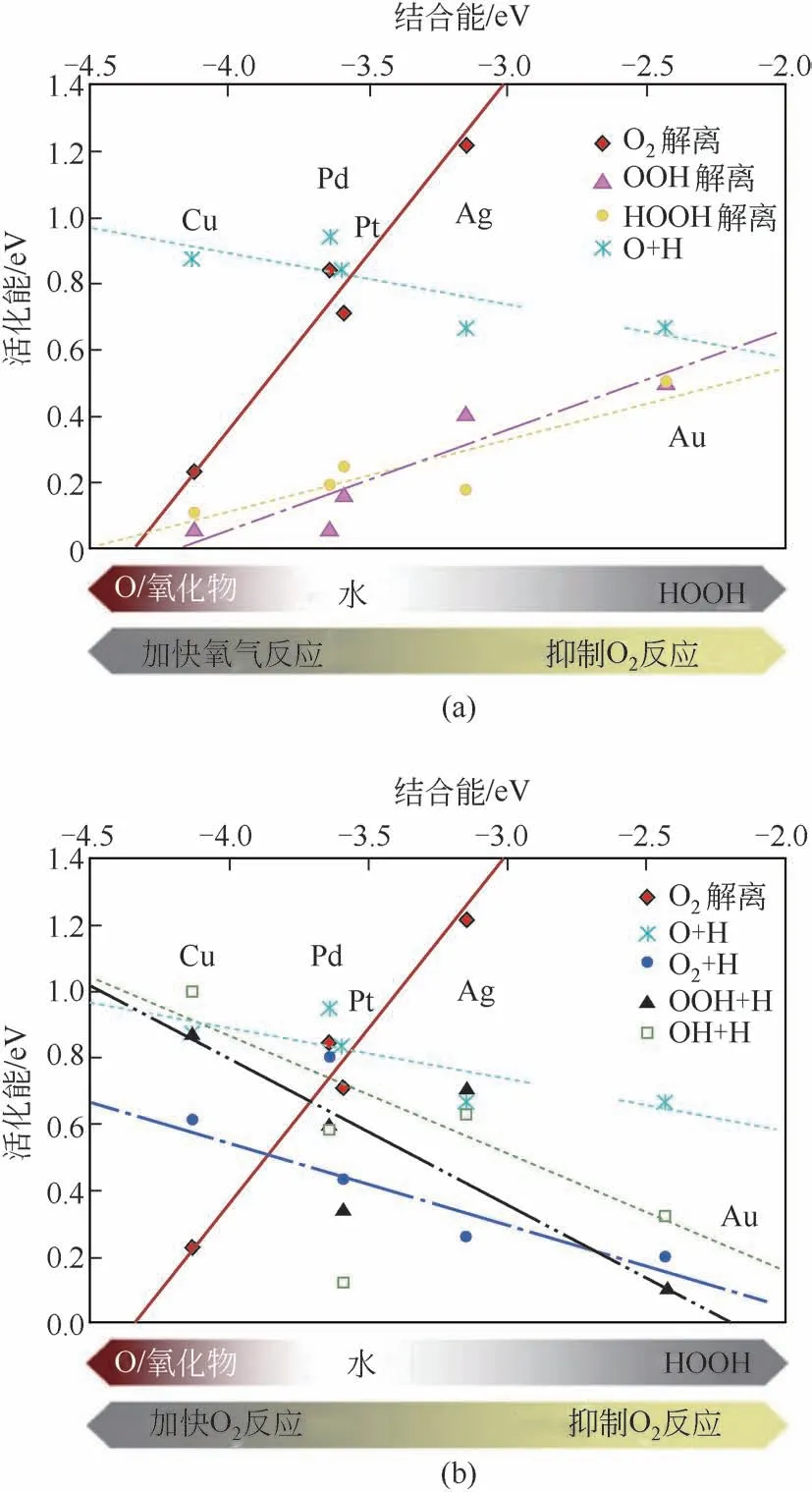
圖2 不同貴金屬催化合成過氧化氫解離活化能[6]
對于金屬Pd直接合成過氧化氫,普遍認為Pd0對于H2和O2有著較強的斷鍵能力,是反應(yīng)的活性位點。然而,Pd0含量并非越高直接合成過氧化氫催化性能越好,Han 等[8]制備Pd/TiO2催化劑直接合成過氧化氫,在不改變粒徑(直徑約為2.4nm)的情況下通過Pd負載量(1%~5%Pd,質(zhì)量分?jǐn)?shù),下同)精確調(diào)整改變Pd 顆粒結(jié)構(gòu)性質(zhì),實驗表明1.0% Pd 催化效果最佳,其單質(zhì)Pd 含量為52.4%,H2O2選擇性達到了61%,比5.0%Pd(單質(zhì)Pd含量64.8%)選擇性高了20%,他們通過DFT 計算分析認為直接合成過氧化氫的活性位點是Pd-PdO 界面(見圖3),不完全還原的Pd 顆粒中存在Pd-O-Pd結(jié)構(gòu),與Pd0顆粒中的Pd-Pd 結(jié)構(gòu)相比Pd-O-Pd 結(jié)構(gòu)對O2和H2O2的解離活化能更高,使得不完全還原的Pd顆粒與完全還原的Pd顆粒相比合成H2O2選擇性提高。

圖3 Pd-PdO界面直接合成過氧化氫機理[8](1?=0.1nm)
2.3 雙金屬催化直接合成過氧化氫
在Pd基催化劑上引入雙金屬如Au等,可以有效調(diào)變Pd顆粒表面的電荷密度,抑制Pd金屬顆粒對O2和H2O2的解離活化,從而提高H2O2選擇性。常見的用于直接合成過氧化氫的雙金屬有Pd-Au、Pd-Pt、Ru-Pd、Ru-Au等[9-12]。
Li等[13]研究了Au-Pd負載于微孔分子篩上的催化直接合成H2O2活性,實驗表明2.5%Au-1.8%Pd/HZSM-5 的反應(yīng)產(chǎn)率達到了138.3mmol/(gcat·h),遠高 于4.3% Au/HZSM-5 的 產(chǎn) 率[4.73mmol/(gcat·h)]。Edwards 等[14]制備了Au-Pd 雙金屬負載于TiO2上用于合成H2O2,實驗表明2.5%Au-2.5%Pd/TiO2的反應(yīng) 產(chǎn) 率 為64mmol/(gcat·h), 高 于5% Pd/TiO2的31mmol/(gcat·h),證實了Au-Pd 雙金屬催化劑具有更高的催化產(chǎn)率。Nugraha 等[15]通過DFT 理論計算分析了Pd-Hg 合金催化選擇性比Pd 更高的原因,DFT 計算表明O2吸附可以發(fā)生在超氧和過氧通道,而與Pd 相比Pd-Hg 具有更多的超氧通道;Pd6Hg3/Pd(111)中的Hg使得其具有與Pd(111)不同的電子表面結(jié)構(gòu)和更低的O2吸附能;由于Pd-Hg 表面Hg 的存在帶來的幾何和電子效應(yīng),Pd 金屬表面與O2具有很強的表面相互作用,不易斷鍵,所以Pd-Hg合金應(yīng)用于直接合成過氧化氫具有更高的選擇性。
Hutchings等[16]合成了不同比例的Au-Pd-Pt催化劑,得到三種合金的活性三相圖(見圖4)。研究表明,當(dāng)比例為2.28%Au-2.28%Pd-0.45%Pt/CeO2時,催化劑具有最高的反應(yīng)產(chǎn)率[670mmol/(gcat·h)]。總的來說,雙金屬或多金屬合金催化劑相對于單金屬催化劑來說有更高的反應(yīng)選擇性,這對于多種金屬催化劑用于直接合成過氧化氫的研究具有指導(dǎo)意義。
在Pd 基催化劑的基礎(chǔ)上引入其他相對惰性的非貴金屬(如Te、Sn 等),也可以有效降低Pd 顆粒邊緣角落位點的電子云密度,提高O2和H2O2在其上的解離活化能,從而抑制O2和H2O2解離等副反應(yīng)提高H2O2選擇性。Hutchings 等[17]制備了不同比例的Pd-Sn/SiO2雙金屬催化劑,并對還原后的催化劑進行了二次氧化,分析測試了還原和氧化前后不同催化劑對于直接合成過氧化氫的反應(yīng)活性(見圖5)。實驗結(jié)果表明,2.5%Pd-2.5%Sn/SiO2與單金屬Pd 基催化劑相比H2O2產(chǎn)率顯著更高,說明Pd、Sn 之間存在協(xié)同效應(yīng);二次氧化后的催化劑與一次氧化催化劑相比,H2O2產(chǎn)率仍穩(wěn)定在較高水平,而H2O2氫化率降至接近于0,H2O2的選擇性最高為95%,說明催化劑中添加適量Sn 產(chǎn)生了顯著的協(xié)同效應(yīng),抑制了H2O2中O—O鍵的斷裂。該作者認為,循環(huán)氧化后的催化劑傾向于生成SnO2包封Pd顆粒的結(jié)構(gòu),使得Pd表面原子電荷密度降低(即配位數(shù)升高),有利于提高H2O2選擇性。在此基礎(chǔ)上,Han 等[18]通過制備Pd-Te 雙金屬催化劑并采用DFT理論計算分析了Pd-Te雙金屬顆粒的反應(yīng)能壘,印證了這一看法。高配位的Pd-Te活性位點與低配位飽和度的Pd 顆粒邊角位相比,其O2解離活化的反應(yīng)能壘更高,對催化劑直接合成過氧化氫的測試也證實了這一計算結(jié)果。
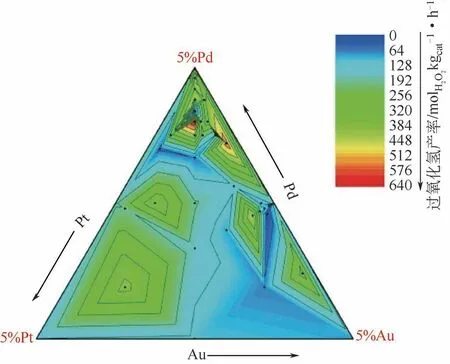
圖4 不同組成的Au-Pd-Pt三金屬催化劑反應(yīng)活性[16]
Pizzutilo 等[19]利用電化學(xué)的方法對Au-Pd 催化劑進行了改性,通過施加不同的電壓,使得Au-Pd顆粒表面的Pd和Au原子以不同的速率溶解在溶液中,得到了不同結(jié)構(gòu)的Au-Pd 顆粒。隨著電壓的升高,金屬表面首先開始解離出Pd,Au 的表面含量增加,再增加電壓,Au、Pd 同時析出,但是析出數(shù)量上Pd>Au,所以金屬會形成Au 殼(如圖6所示)。實驗表明施加電壓為1.6V 時,Au-Pd 顆粒表面形成了Au 包裹的Au-Pd 顆粒,此時直接合成過氧化氫的選擇性達到了最高(85%)。
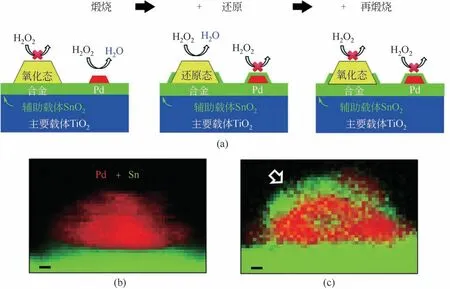
圖5 Pd-Sn/SiO2多次氧化還原反應(yīng)機理(a)、合金煅燒前后Pd、Sn分布對比[(b)、(c)][17]
目前大部分雙金屬催化劑采用制備策略為同時負載法或覆蓋法,前者難以有效覆蓋Pd顆粒邊緣角落位點,后者覆蓋了大部分活性位點使得反應(yīng)活性急劇降低。Liang等[20]合成了納米片層狀的HZSM-5分子篩,隨后按不同順序負載1.5%Pd和0.5%Au,得到了不同結(jié)構(gòu)的納米片層狀金屬催化劑(見圖7)。將制備的不同雙金屬催化劑用于直接合成過氧化氫,分析不同的金屬結(jié)構(gòu)對于合成H2O2催化性能的影響。在分子篩限域作用下從納米片載體片層間引入雙金屬Au得到雙金屬納米片催化劑Au-Pd/ZN,實現(xiàn)區(qū)域選擇性覆蓋Pd納米片邊緣角落位點,可進一步抑制副反應(yīng)。實驗發(fā)現(xiàn),與同時負載的AuPd納米片催化劑AuPd/ZN 相比,在相同反應(yīng)條件下,Au-Pd/ZN 的反應(yīng)轉(zhuǎn)化率較AuPd/ZN 低20%,Au-Pd/ZN 的H2O2選擇性相較AuPd/ZN 提高了40.4%,Au-Pd/ZN表現(xiàn)出更高的反應(yīng)產(chǎn)率。

圖6 電化學(xué)溶析制備的Au-Pd催化劑[20]
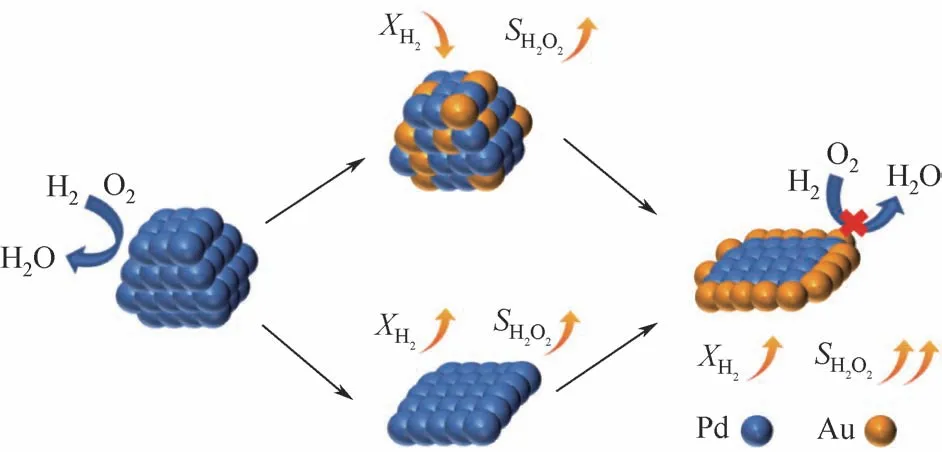
圖7 邊角位點覆蓋Au的雙金屬AuPd催化劑[20]
3 直接合成過氧化氫催化劑用載體
對于直接合成過氧化氫來說,載體的性質(zhì)是一個關(guān)鍵因素。在之前的報道中,使用的載體材料有分子篩[21]、金屬氧化物[22]、碳材料[23]、SiO2[24]、雜多酸[25]、膜材料[26]等。
3.1 載體酸性
在反應(yīng)過程中,反應(yīng)體系的酸性對于直接合成過氧化氫的性能有顯著影響。質(zhì)子酸的存在可以抑制H2O2氫化和直接分解,從而提高反應(yīng)的選擇性[27]。同時,H+的存在可以增強反應(yīng)的活性,Wilson 等[28]提出了一種類似二電子氧還原(ORR)的理論,認為H+參與了直接合成過氧化氫的中間步驟,可以提高過氧化氫產(chǎn)率(見圖8)。他們認為在Pd金屬內(nèi)部存在電子轉(zhuǎn)移通道。H2在Pd顆粒表面吸附斷鍵,氧化生成H+擴散到溶液中;H2氧化生成的電子e-在Pd 顆粒中通過質(zhì)電通道轉(zhuǎn)移;O2吸附于Pd顆粒表面接受e-還原生成O2-2 后與溶液中H+結(jié)合生成H2O2并脫附。該文通過改變?nèi)軇┲蠬+濃度,發(fā)現(xiàn)質(zhì)子對H2O2生成是重要的。然而目前為止還沒有進一步的工作驗證該理論。
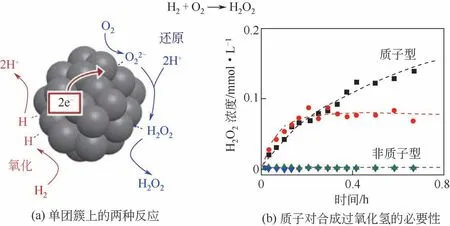
圖8 氫離子在合成過氧化氫中的作用[28]
研究中常在溶劑中添加無機酸以提高反應(yīng)的選擇性和活性,但是無機酸的引入會造成活性金屬浸出流失、反應(yīng)器腐蝕等問題,使用酸性載體替代無機酸是目前直接合成過氧化氫研究方向之一。Park等[29]制備了一種SO3H官能化的SiO2載體并負載Pd。實驗發(fā)現(xiàn),SO3H 官能團的引入不僅提高了H2O2的收率,還使得在反應(yīng)過程中不使用無機酸,避免了金屬的流失等問題。Kim 等[30]將樹脂K2621 接枝含硫烷基長鏈后,離子交換制備SO3H 官能化樹脂后負載Pd 得到Pd/K2621,在持續(xù)的半間歇反應(yīng)中最后得到了8.9%的H2O2,產(chǎn)率為180g H2O2/(gPd·h)。另外一種常見的提高載體酸性的方式是直接使用酸性載體。Hutchings 等[31]制備了Cs 交換的磷鎢酸CsxH3-xPW12O40作為固體酸助劑,探究了多種不同酸添加劑對Au-Pd/TiO2(2.5% Au-2.5% Pd,質(zhì)量分?jǐn)?shù)) 的直接合成H2O2性能的影響,發(fā)現(xiàn)ZrO2、HZSM-5、CsxH3-xPW12O40幾種助劑能夠有效地提高H2O2的產(chǎn)率,這一研究可有效地指導(dǎo)選擇直接合成H2O2的載體。
3.2 載體表面性質(zhì)
除載體酸性外,載體的其他表面性質(zhì)對直接合成過氧化氫也存在一定影響,如含氧官能團、親疏水性等。Gudarzi 等[32]研究了含氧基團對影響H2O2催化分解的影響,發(fā)現(xiàn)碳載體上含氧表面官能團可以穩(wěn)定溶液中的H2O2,減少其分解和氫化反應(yīng)。Blanco 等[33]通過在SiO2表面接枝不同酸性基團發(fā)現(xiàn),接枝羧基Si-COOH 的H2O2選擇性為22%,比接枝磺酸基團的Si-SO3H 選擇性(55%)顯著更低,分析認為Pd 顆粒與磺酸基團的相互作用更強使得反應(yīng)選擇性更高。此外,載體的親疏水性對反應(yīng)的影響也不可忽略。直接合成過氧化氫反應(yīng)過程中氣相通過液相體系到達催化劑活性中心,而研究表明溶劑對氫氣和氧氣的溶解度很大程度決定了反應(yīng)產(chǎn)率,證明反應(yīng)受傳質(zhì)影響嚴(yán)重[34]。
Giacomo 等[35]利用十六烷基-2-羥乙基-二甲基磷酸二氫銨(HHDMA)作為Pd納米粒子的封裝劑(見圖9)制備了Pd-HHDMA/TiO2用于直接合成過氧化氫。隨著HHDMA的加入直接合成H2O2選擇性由10% 提升至80%,且與聚乙烯封裝相比,HHDMA能夠穩(wěn)定包裹在Pd顆粒外,而聚乙烯隨著反應(yīng)進行有機物流失嚴(yán)重;DFT分析表明,反應(yīng)形成的中間體H2O2*在金屬表面吸附時,在沒有配體的情況下,H2O2*與Pd吸附是相對自由的,而有配體時由于配體的靜電相互作用,會使H2O2*成鍵呈垂直狀,可以有效地提高副反應(yīng)的活化能,防止H2O2*與表面接觸而分解或者過度氫化。為此,對催化劑金屬進行針對性的修改改性,可加快反應(yīng)的擴散或提高Pd顆粒的O—O鍵解離活化能,對于提高反應(yīng)活性和H2O2選擇性具有重要意義。
3.3 載體摻雜物
催化劑載體中的摻雜物也會影響直接合成過氧化氫反應(yīng)。Abate 等[36]研究了N 摻雜的碳納米管負載金屬Pd 和Au-Pd,發(fā)現(xiàn)N 摻雜的納米管載體能夠有效的提高Pd 納米顆粒的分散度和穩(wěn)定性,提高H2O2的收率,此外N摻雜的基團的引入還能提高催化劑表面的酸性,增強H2O2的穩(wěn)定性。Hutchings 等[25]利用雜多酸載體摻雜金屬陽離子(Cs+、Rb+、K+、Ag+),發(fā)現(xiàn)含Cs+、Rb+的催化劑能有效提高H2O2反應(yīng)活性。
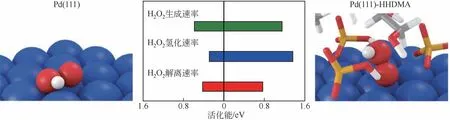
圖9 烷基長鏈作用機理圖[35]
3.4 載體結(jié)構(gòu)形貌
載體的結(jié)構(gòu)形貌對催化劑直接合成過氧化氫的性能也會有影響。Song 等[40]通過將Pd 顆粒負載在兩種不同的介孔硅分子篩MCM-41 和SBA-15 上,分析了介孔孔徑和壁厚對于催化劑性能的影響[見圖10(a)]。分子篩MCM-41介孔孔徑較窄(2.9nm),負載金屬后得到一部分小Pd 納米顆粒(通道中約2nm)和一部分大Pd 顆粒(位于支撐通道外部,數(shù)量少但質(zhì)量大)。小Pd納米顆粒表現(xiàn)出極強的反應(yīng)活性,但是對應(yīng)的選擇性很低,此外MCM-41的薄壁厚度也使得催化穩(wěn)定性存在一定問題。分子篩SBA-15介孔孔徑較大(8.4nm),負載得到的Pd納米粒子90%處于孔道內(nèi),平均粒徑在4.5nm 左右,高分散性使得反應(yīng)活性較高,與負載MCM-41的催化劑相比反應(yīng)選擇性也較高。同時,SBA-15 的適宜的孔徑使得溶劑和產(chǎn)物容易擴散,SBA-15 負載型催化劑的較厚壁厚也使得催化劑具有良好的機械穩(wěn)定性和可重復(fù)使用性。
針對載體結(jié)構(gòu)的特異性設(shè)計有助于提高直接合成過氧化氫的催化性能。Kim 等[41-42]制備了一種球形SiO2包裹Pd 顆粒的結(jié)構(gòu)Pd@SiO2,[見圖10(b)],表征發(fā)現(xiàn)該載體負載的金屬具有良好的分散性(分散度43%),均一的粒徑(約4nm),實驗表明Pd@SiO2與Pd/SiO2和Pd/Al2O3相比具有很高的反應(yīng)活性,說明通過核殼結(jié)構(gòu)提高少配位的Pd 顆粒分散度,進而提高H2O2產(chǎn)率是有效的。Kim等[43]還制備了中空的核殼結(jié)構(gòu)催化劑Pd@Void@ZrO2,實驗發(fā)現(xiàn)Pd@Void@ZrO2的活性為Pd@SiO2的1.2 倍。Kim 等[43]制備了一種介孔SiO2殼包封的Pd-納米晶體接枝的SiO2的納米珠[見圖10(c)],在活性測試中殼層越薄直接合成H2O2反應(yīng)活性越好,但殼層厚度應(yīng)適當(dāng),否則殼層太薄會使Pd 顆粒在煅燒還原過程中發(fā)生團聚。實驗表明,具有介孔的M(2)活性約為微孔S(2)的4倍。
3.5 金屬載體相互作用
不同載體的催化劑直接合成過氧化氫的活性不同也可能是受到金屬和載體間的相互作用的影響。Park 等[44]合成了Pd/SiO2和Pd/TiO2用于直接合成過氧化氫,實驗發(fā)現(xiàn)Pd/TiO2的選擇性比Pd/SiO2高,而活性更弱,因為Pd 納米顆粒與TiO2形成的Pd-O-Ti 相互作用強于Pd-O-Si,使得Pd/TiO2中Pd 表面的電荷更低,電荷密度越低其金屬顆粒對反應(yīng)物的斷鍵能力越弱,使得活性較低而選擇性升高。
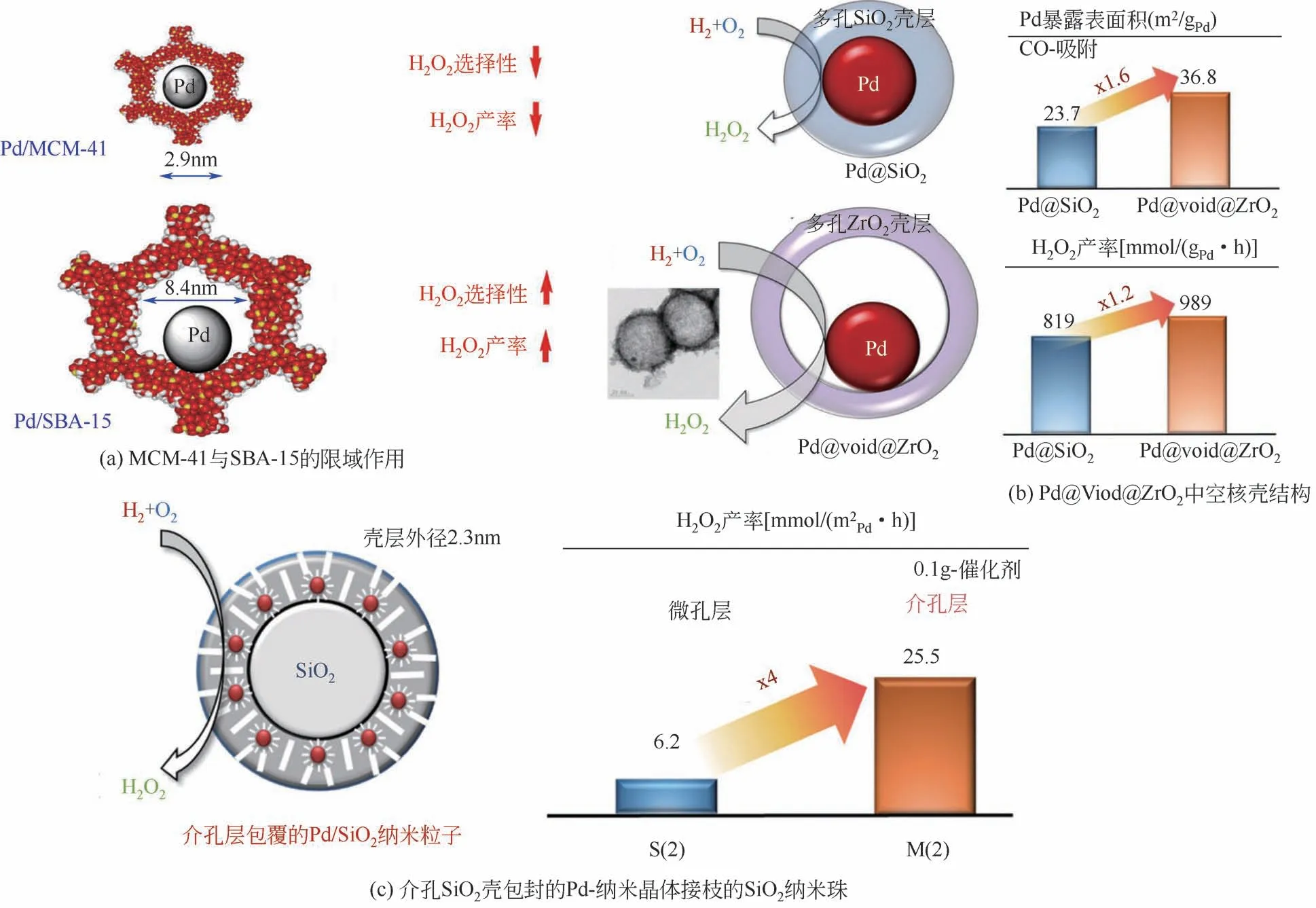
圖10 不同載體形貌對反應(yīng)的影響[40-41,43]
Torrente-Murciano 等[45]合成了嵌入鈦酸鹽納米管中的Au-Pd 催化劑0.4% Au-1.5% Pd/Ti-NT,其H2O2產(chǎn)率達到了11600mmol/(gPd·h),而對應(yīng)的負載型催化劑1.5% Au-1.5% Pd/TiO2的產(chǎn)率僅為430mmol/(gPd·h)。該作者認為納米管表面的曲率和化學(xué)環(huán)境對于Au-Pd納米顆粒的形態(tài)和穩(wěn)定性起到了關(guān)鍵作用,納米管中高分散的Au-Pd 顆粒(<2nm)與鈦酸鹽載體間存在著較強的載體和金屬相互作用,使得反應(yīng)活性與選擇性均較高。
對于分子篩載體來說,載體酸性位(B 酸位)與金屬顆粒在煅燒的過程中結(jié)合,形成Pd-O-Si/Al的結(jié)構(gòu),該結(jié)構(gòu)可以有效的改變金屬顆粒表面的電子結(jié)構(gòu)。Liang等[5]通過制備不同硅鋁比納米片分子篩載體(nanosheet HZSM-5)的Pd 催化劑,分析了載體酸性對金屬載體相互作用的影響,發(fā)現(xiàn)載體酸性位點使得Pd 的XPS 和CO-FTIR 表征特征峰發(fā)生了不同程度的偏移,表明載體酸性可以有效改變金屬的電子結(jié)構(gòu),測試直接合成過氧化氫催化性能發(fā)現(xiàn)Pd 顆粒表面電荷密度越低,催化選擇性越高(見圖11)。
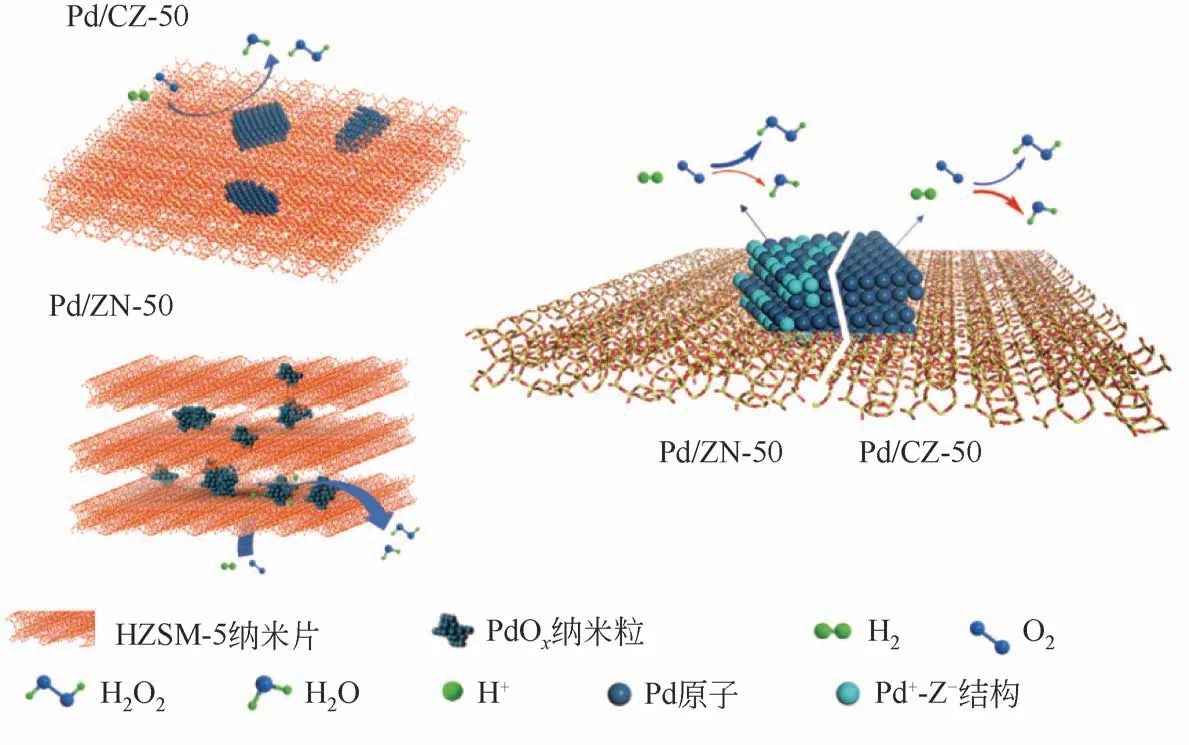
圖11 納米片分子篩催化劑的金屬載體相互作用[10]
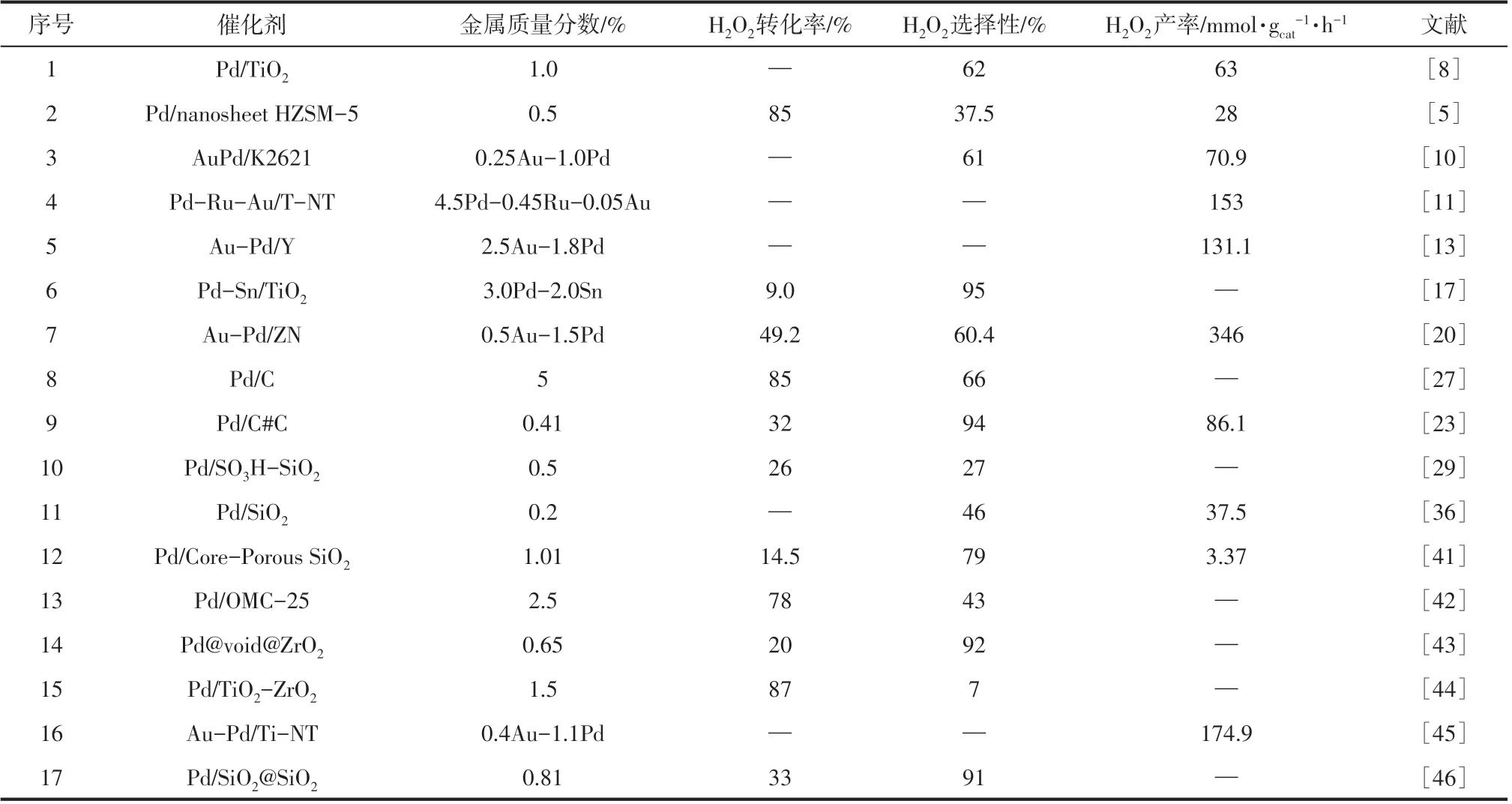
表2 近年來各催化劑直接合成過氧化氫催化性能
總結(jié)近年來的各催化劑催化性能(見表2)可看出,目前用于直接合成過氧化氫的催化劑產(chǎn)率普遍較低,仍難以滿足工業(yè)化的要求。
4 結(jié)語
目前,氫氧催化直接合成過氧化氫研究取得了許多重大進展,主要集中在催化機理、催化活性中心、催化載體及改性等方面。過氧化氫合成過程中的活性和選擇性與各反應(yīng)物及反應(yīng)產(chǎn)物的吸附/脫附能和生成/解離活化能相關(guān),直接合成過氧化氫選擇性提高的關(guān)鍵在于提高O2和H2O2的解離活化能,抑制O—O 鍵的斷裂。通過改變金屬顆粒尺寸、引入雙金屬、增強金屬與載體相互作用等方式調(diào)變活性中心的解離活化能力,能夠抑制O2和H2O2的解離活化,提高反應(yīng)的選擇性。氫氧直接合成過氧化氫作為一種綠色合成過氧化氫的方法,目前研究大多仍處于實驗階段,研究和設(shè)計高活性選擇性的催化劑和適宜的反應(yīng)體系提高反應(yīng)產(chǎn)率,對于未來直接合成過氧化氫的工業(yè)化應(yīng)用有著重要的意義。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
