含磷多孔有機聚合物的合成及其在多相催化中的應用
2021-04-20 10:30:26吳淼江孫鵬李福偉
化工進展 2021年4期
關鍵詞:催化劑
吳淼江,孫鵬,李福偉
(1 中國科學院蘭州化學物理研究所羰基合成與選擇氧化國家重點實驗室,甘肅蘭州730000;2 中國科學院大學,北京100049)
膦配體(本文指三價有機膦類、亞磷酸酯類或亞磷酰胺類配體)既是σ電子給體也是π電子受體(圖1)[1],作為一種“軟堿”與后過渡金屬釕[2]、鋨[3]、鈷[4]、銠[5]、銥[6]、鎳[7]、鈀[8]、鉑[9]等有著很好的配位能力。金屬-膦配合物作為均相催化劑在碳氫鍵、飽和碳碳鍵、不飽和碳碳鍵、碳氮鍵、碳氧鍵、碳鹵鍵等的活化或構建中有著廣泛的應用,而且在反應中常常能表現出很高的催化活性和選擇性。但它們溶解于反應體系中難以分離回收,大大限制了其在大規模生產中的應用。

圖1 有機膦配體的電子性質
為了方便金屬-膦配合物催化劑的回收和重復利用,科研人員嘗試將膦配體錨定在有機高分子載體或無機載體上,制備錨定的金屬-膦配體催化劑(圖2)。其中,用可溶性聚合物錨定膦配體是具有代表性的方法之一。可溶性聚合物能溶于反應體系,與均相催化劑極為接近,官能團化的膦配體很容易通過接枝或共聚的方式引入,因此20 世紀末報道了大量的可溶性聚合物基金屬-膦配體催化劑。常見的用于錨定膦配體的可溶性聚合物有線性聚苯乙烯[圖2(a)][10]、線性聚乙二醇[圖2(b)][10]、肽鏈聚合物[圖2(c)][10]以及支鏈聚合物[圖2(d)][11]等。但可溶性聚合物基催化劑需要通過添加沉淀劑、微濾或超濾等手段來實現催化劑的回收,且小分子聚合物鏈有可能殘留于產物中產生污染,因此逐漸被研究人員所舍棄。隨著無機-有機雜化材料的興起,科研人員將官能團化的膦配體通過吸附或者化學鍵合的方式與無機材料結合制備含磷多相催化劑。其中,具有代表性的無機材料有高比表面積的有序介孔硅材料[圖2(e)、(f)][12]和具有磁性的Fe3O4納米顆粒[圖2(g)][13]。但是無機材料表面活性位點單一,化學修飾潛力十分有限,大大限制了基于膦配體-無機載體催化劑的開發。
多孔有機聚合物(porous organic polymers,POPs)是由有機小分子通過共價鍵聯結而成的孔基材料。相較于可溶性聚合物,多孔有機聚合物具有比表面積大、孔隙發達和易于從溶劑體系中分離的優勢;相較于有機-無機雜化材料,它具有分子骨架穩定、可調變性和可修飾性強的優勢。因此,多孔有機聚合物在分離、多相催化、污染物捕集、氣體儲存等方面有著廣泛的應用前景[14]。

圖2 傳統方式錨定膦配體示例
多孔有機聚合物中微孔、介孔和大孔往往同時存在,這種多級孔的特征特別適合于用作多相催化劑載體[15],并能結合均相催化體系的某些優勢[16]。由于多孔有機聚合物的結構易于調變,其在仿生催化材料方面具有開發潛力[17]。迄今為止,雖已有一些關于多孔有機聚合物的制備及其應用的綜述發表[14,18-19],但還沒有概述和總結含磷多孔有機聚合物及其在多相催化中應用的綜述發表(本文所述聚合物單體包含三價有機膦類、亞磷酸酯類、磷雜苯類、氯化磷類和季鹽類,因此用意含磷元素的“磷”命名聚合物,即“含磷多孔有機聚合物”)。事實上,作為多相催化劑制備新平臺,近十年來含磷多孔有機聚合物載體的發展十分迅速,甚至在工業化生產中已有應用。中國科學院大連化學物理研究所丁云杰和嚴麗團隊[20]基于含磷多孔聚合物基催化技術,實現了乙烯氫甲酰化制備丙醛/正丙醇的多相催化,推動5 萬噸/年的制備丙醛/正丙醇工業裝置成功投產。研究人員在膦配體單體設計、聚合物合成方法等方面做了大量的工作,含磷多孔有機聚合物已成為一個有活力且極具發展潛力的領域。因此,很有必要對近十年來含磷多孔有機聚合物基催化劑的制備和應用進行系統性整理。結合本文作者課題組在卡賓聚合物基催化劑方面的理解[21-25],本文對含磷多孔聚合物基催化劑設計、合成以及應用過程中發現的一些規律進行歸納和闡述。

圖3 含磷多孔有機聚合物合成方法
目前為止,文獻報道的含磷多孔有機聚合物制備方法有偶聯縮聚、鋰鹽參與的縮聚、Friedel-Crafts 縮聚、溶劑熱烯烴聚合、Scholl 縮聚、酚醛聚合、醛胺縮聚、聚吡喃鹽的磷代以及多段式聚合等(圖3)。多元的聚合方法意味著聚合單體結構的多樣化,使得聚合單體的設計和開發十分靈活多變。而合成方法的多樣性以及合成單體的多變性又使得含磷多孔有機聚合物具有巨大的調變空間,可以根據各種催化反應體系的特點有針對性、有目的性地設計和調變催化劑結構。這使得制備高活性、高選擇性的催化劑成為可能。可調變性和可修飾性強是含磷多孔有機聚合物催化劑的優勢,同時也是此類催化劑發展面臨的難點和挑戰:如何在如此多變的體系中找到適合反應體系的催化劑?圍繞這一核心問題,本文對近十年來含磷多孔有機聚合物載體的制備方法、聚合單體進行了總結,旨在為含磷多孔有機聚合催化劑的設計合成提供些許指導和幫助,進一步加快此類催化劑的發展及工業應用進程。
多孔有機聚合物的分類和命名十分繁雜,如微孔有機聚合物(microporous organic polymers,MOPs)、自具微孔聚合物(polymers of intrinsic microporosity, PIMs) 、 超 交 聯 聚 合 物(hypercrosslinked polymers,HCPs)、共軛微孔聚合物(conjugated microporous polymers,CMPs)、共價有機框架(covalent organic frameworks,COFs)、編織芳基網絡聚合物(knitting aryl network polymers,KAPs)、多孔離子聚合物(porous Ionic Polymers,PIPs)等。在某些情況下,各種方式命名的多孔聚合物之間不能明確地界定。為使敘述更加清晰明了,本文以制備方法進行分類來對含磷多孔有機聚合物進行綜述。
1 偶聯縮聚
偶聯縮聚是具有多反應位點的單體間通過偶聯形成碳碳鍵交聯在一起,是制備多孔聚合物的常用方法,這類反應包括Heck 偶聯、Sonogashira-Hagihara 偶聯、Suzuki-Miyaura 偶聯和Yamamoto 偶聯等(圖4)[26]。基于此,一系列具有多反應位點的膦配體單體和共聚單體被合成,并用于含磷多孔有機聚合物的制備。
1.1 Heck偶聯縮聚
Heck 偶聯縮聚是利用多鹵代芳烴單體和多乙烯基芳烴單體間的交叉偶聯,進而高度交聯成聚合物的方法。2015年,張帆團隊[27]分別用膦配體單體a1、a2和共聚單體b1,通過Heck偶聯縮合“一鍋”合成了含磷多孔有機聚合物基鈀納米顆粒催化劑。該催化劑在Suzuki-Miyaura 偶聯反應中表現出很好的活性,分別基于a1 和a2 的聚合物催化劑在Suzuki-Miyaura 偶聯反應中表現出相似的催化活性,體系中磷的價態對鈀物種催化活性的影響較小。值得注意的是,該文還指出在一定范圍內偶聯縮聚鈀催化劑用量增加能促進縮聚反應的進行,從而使得聚合物孔徑減小、比表面積增大。但鈀催化劑用量過高,又可能因鈀顆粒占據或堵塞孔結構導致比表面積減少。2017年,詹莊平團隊[28]使用乙烯基化的膦配體單體a3和溴代單體b2,經Heck偶聯縮合、碳碳雙鍵還原兩步成功合成了含磷多孔聚合物。由于骨架結構中碳碳雙鍵被還原成了更具柔性的單鍵,該聚合物孔徑較上述張帆團隊合成的聚合物具有更大的孔徑。在多相催化中,較大的孔道有利于底物在催化劑中的擴散,從而更快地接觸到活性位點。該聚合物負載鈀納米顆粒后,在硝基芳烴、α,β-不飽和烯烴等化合物的加氫反應中表現出高活性、好的底物適用性和重復使用性。

圖4 偶聯反應
1.2 Sonogashira-Hagihara偶聯縮聚
為了防止炔烴單體間的自身偶聯,Sonogashira-Hagihara偶聯縮聚必須在嚴格無氧的條件下進行。2015 年,李燦團隊[29]使用(R)-4,4-DibromoBINAPO 單體(a4),分別同b3、b4、b5 和b6 通過Sonogashira-Hagihara 偶聯縮聚、三氯硅烷脫氧兩步成功合成了一系列含BINAP 的聚合物載體。實驗結果表明,共聚單體對催化劑性能影響明顯:基于共聚單體b6 的聚合物與[RuCl2(benzene)]2配位后在β-酮酯的不對稱加氫反應中表現出很好的活性和手性選擇性,轉化率和對映體過量值(enantiomeric excess,ee) 均能達到99%;其與[Ir(COD)Cl2]配位后,在喹哪啶的不對稱加氫中表現出比均相催化體系(BINAP/[Ir(COD)Cl]2)更高的催化活性和相當的選擇性。該項工作說明通過膦配體單體調控聚合物基催化劑活性和選擇性是可行的。
1.3 Suzuki-Miyaura偶聯縮聚
2017 年,李濤團隊[30]用a5 和b7 通過Suzuki-Miyaura 偶聯縮聚“一鍋”合成了聚合物基鈀納米顆粒催化劑,在水和乙醇的混合溶劑中對Suzuki-Miyaura 偶聯反應表現出很好的催化活性和良好的底物適用性。該催化劑重復使用5次后,XPS譜圖中零價鈀比例顯著上升,說明鈀發生了團聚。2019年,Bojdys 團隊[31]通過Suzuki-Miyaura 偶聯縮聚,使用甲氧基穩定的λ5磷雜苯類化合物a6 和b7 成功合成了π 共軛的λ5磷雜苯共價網絡聚合物(covalent phosphinine framework,CPFs)。以三乙醇胺為犧牲劑,在波長380~780nm的可見光條件下,該聚合物在鈀(合成過程中殘留)的助催化下光解析氫效率速率為33.3mmol/(h·g)。要拓展磷雜苯類聚合在多相催化中的應用,必須要合成比λ5磷雜苯具有更強配位能力的λ3磷雜苯聚合物。但實際上,甲氧基穩定的λ5磷雜苯單體結構也很容易被酸、堿破壞,而λ3磷雜苯是更不穩定的單體分子。因此,從λ3磷雜苯小分子單體出發合成含磷雜苯的聚合物是很困難的。
1.4 Yamamoto偶聯縮聚
2012 年,張所波團隊[32]使用季鹽單體a8 通過Yamamoto 自偶聯縮聚制得了季鹽型多孔有機聚合物。該聚合物直接使用能催化環氧化合物與二氧化碳生成環碳酸酯的反應,負載鈀納米顆粒后能用于催化Suzuki-Miyaura 偶聯反應。次年,他們[33]又用a5、a7分別合成了兩種比表面積超過1200m2/g的多孔聚合物,該聚合物500℃條件下還能保持穩定。通過濃鹽酸處理和溶劑洗滌的方式除去鎳金屬縮聚催化劑,再引入鈀物種制成聚合物基鈀納米催化劑,能催化Suzuki-Miyaura 偶聯反應的進行。與前文張帆團隊[27]的實驗結果不同,磷物種價態對張所波團隊制備的催化劑活性有著明顯影響:基于三價膦單體(a5)的聚合物催化劑活性明顯高于基于五價的氧磷單體(a7)的聚合物催化劑,說明三價膦配體促進了鈀物種在該反應體系中的催化活性。
近十年來通過偶聯縮聚制備的含磷多孔有機聚合物,其單體組分、物理性質(比表面積、孔體積和孔徑)、負載金屬類型以及相應催化劑的應用參見圖5和表1。

圖5 偶聯縮聚單體

表1 偶聯縮聚含磷聚合物單體組成、孔結構參數及其在多相催化中的應用
前文例舉的一些文獻結果表明,含磷單體和共聚單體均對聚合物物理性質(比表面積、孔體積、孔徑分布等)有影響,且與聚合物基催化劑催化性能密切相關。縮聚催化劑的量也能影響聚合物物理性質。催化劑量越大越有利于縮聚,一定范圍內隨縮聚催化劑量增加,合成的聚合物孔徑變小、比表面積增大[27]。由表1可知,通過偶聯縮聚合成的含磷多孔有機聚合物孔徑分布普遍較窄,主要由微孔和孔徑偏小的介孔組成[參照國際純粹與應用化學聯合會(IUPAC)對多孔材料的分類:微孔<2nm、介孔2~50nm、大孔>50nm],說明偶聯縮聚制得的聚合物高度交聯。值得注意的是,不同偶聯縮聚方式合成聚合物的性質也有所差別。Suzuki-Miyaura和Yamamoto偶聯縮聚合成的含磷多孔有機聚合物,單體之間由苯環間的碳碳鍵直接相連,具有很強的剛性。而Heck、Sonogashira-Hagihara 偶聯縮聚合成的多孔聚合物中的碳碳雙建和三鍵還原后變成柔性的烷基鏈段,相比Suzuki-Miyaura 和Yamamoto偶聯縮聚理論上能得到孔徑更大、溶脹性更好的聚合 物。 此 外, Heck、 Sonogashira-Hagihara 和Suzuki-Miyaura 偶聯縮聚需要鈀催化劑的參與,Yamamoto 偶聯縮聚需要鎳催化劑的參與,而鈀和鎳都能與膦配體配位并且會在催化過程中團聚,這勢必給負載其他金屬帶來麻煩。從表1 可以看出,通過偶聯縮聚制備的聚合物基催化劑負載金屬大部分是鈀,單一的金屬負載一定程度上限制了其在多相催化領域的應用。如果在鈀或鎳的基礎上再引入其他金屬物種應用在某些特定的領域(如需要多活性位點的串聯催化),不僅能拓寬偶聯縮聚聚合物基催化劑的應用,還能省去去除縮聚催化劑的步驟。
2 鋰鹽參與的縮聚

圖6 鋰鹽參與的縮聚
2008年,Kaskel課題組[34]首次通過二取代聯苯鋰鹽與硅酸乙酯反應制得多孔聚合物。2012 年,他們[35]將該方法用于含磷多孔有機聚合物的合成。如圖6,二溴代聯苯(a9)與正丁基鋰反應生成二鋰代聯苯,再與三氯化磷(b8)偶聯縮聚得到含磷聚合物。該聚合物負載RhCl(PPh3)3后用于環己酮與異丙醇的氫轉移反應,二次使用時活性明顯下降,催化劑穩定性差。2013 年,Bruijnincx 團隊[36]同樣用單體a9 和b8 合成了含磷聚合物,并用苯基鋰進一步除去聚合物中殘留的氯元素。該文指出鈀前體對其在聚合物中的分散形式影響明顯。聚合物負載零價的Pd(dba)2后從電鏡照片觀察到鈀以納米顆粒的形式分散在聚合物中,負載二價的Pd(acac)2則沒有發現以納米顆粒存在的鈀物種,不過以Pd(acac)2為前體制備的催化劑在使用過程中或Pd(acac)2過量的情況下還是會有鈀納米顆粒的生成。該作者還研究了鈀與磷元素的比例對鈀配位方式的影響。磷/鈀比小于2時,13P核磁譜圖中未配位的磷與配位磷的信號峰共存;磷/鈀比接近2 時,13P 核磁譜圖中未配位磷的信號峰基本消失。雖然不能確定鈀與磷是否為二齒配位,但卻證明了該聚合物中幾乎所有磷位點都能與鈀配位,說明該法合成的含磷聚合物中磷位點極少被聚合物骨架包埋。2016 年,Palkovits 團隊[37]通過鋰鹽縮聚的方法制備了用于甲酸分解制氫的聚合物基釕催化劑。該文指出基于雙膦單體a10的釕基聚合物催化劑催化活性和選擇性均優于基于a9和a11的催化劑,重復使用7次后只有少許Ru 納米顆粒出現,具有良好的穩定性。對比以上3個團隊制備的聚合物比表面積(表2),后兩者采用了苯基鋰后處理的縮聚物比表面明顯要低一些,說明苯基取代未參與反應的氯原子的同時會占據相當一部分的孔體積。

表2 鋰鹽縮聚含磷聚合物單體組成、孔結構參數及其在多相催化中的應用
2019 年,Yoon 團隊[38]使用四反應位點的單體b9 同雙膦單體a10 制得了含磷聚合物。相較于b8,b9是一個具有更多反應位點的立體型的交聯單體,制得的聚合物即使經苯基鋰處理,其比表面積和孔體積依然能達到469m2/g和0.475cm3/g。該聚合物引入RuCl3后制得的催化劑在二甲胺的氫甲酰化反應(二氧化碳為羰源)中表現出很好的活性和穩定性。鑒于RuCl2(dppe)2早就被證明是該反應的非常有效的均相催化劑[39-40],而Yoon團隊所制備的聚合物中包含類1,2-雙(二苯基膦)乙烷(dppe)的結構。這說明均相催化理論能對含磷聚合物基催化劑單體的設計和選擇提供方向和指導。
本部分提到的單體、聚合物物理性質以及各聚合物基催化劑的應用匯總見圖7和表2。

圖7 鋰鹽縮聚單體
鋰鹽與氯化磷的縮聚反應比較容易進行且不可逆,理論上會使得單體之間連接的無序度增加。與偶聯縮聚聚合物相比,該類聚合物孔道更大且分布不均勻。表2中聚合物孔徑分布就很好地驗證了這一點,其氮氣吸附等溫線呈典型的多級孔分布(詳見對應參考文獻)。此外,正丁基鋰、苯基鋰和氯化磷對水分極其敏感,反應活性高,使得此類縮聚反應具有一定危險性。
3 Friedel-Crafts縮聚
Friedel-Crafts縮聚是芳香族化合物在路易斯酸的催化下,通過偶聯劑相互聯結形成超交聯網狀無規聚合物的方法。自2012 年譚必恩和李濤團隊[41]首次將該方法應用于合成含磷多孔有機聚合物合成以來,科研人員一直試圖拓展該方法在多相催化領域的應用。Friedel-Crafts縮聚能合成含大孔、介孔和微孔的多級孔含磷多孔聚合物載體,也能通過選擇合適的縮聚單體和偶聯劑制備微孔或介孔含磷多孔聚合物載體。
3.1 膦配體間的Friedel-Crafts縮聚

圖8 單體a12、b10和c1間的Friedel-Crafts縮聚
2012 年,譚必恩團隊用單體a12、b10 和偶聯劑c1 在氯化鐵的催化下首次成功合成了比表面積高達1036m2/g 的多級孔KAPs(Ph-PPh3)[圖8(a)],膦配體均勻分散于KAPs(Ph-PPh3)體相中。該聚合物負載PdCl2后用于鹵代芳烴[41]或氯化芐[42]與苯硼酸類化合物的Suzuki-Miyaura 交叉偶聯反應表現出很高的活性。多級孔載體中大孔和介孔有利于傳質,微孔則有利于金屬的均勻分布和穩定。為了闡述微孔對催化劑穩定性能的作用,他們還使用另一種方式制備聚合物基鈀催化劑作為比較。如圖8(b),該作者先用b10和c1縮聚合成KAPs(Ph),然后再加入含磷單體a12 進行第2 步縮聚合成KAPs(Ph)-PPh3。該法制備的聚合物中膦配體更多的是分散于聚合物表面,兩種KAPs 負載PdCl2后分別記為KAPs(Ph-PPh3) -Pd 和KAPs(Ph) -PPh3-Pd, 用 作Suzuki-Miyaura 交叉偶聯催化劑。實驗表明,后者鈀流失嚴重,使用1次就有鈀黑生成,第2次使用時活性明顯降低;而前者首次使用鈀流失率僅約1%,重復使用5次后催化活性無明顯下降,催化劑電鏡照片與新鮮催化劑沒有明顯變化。這說明微孔能有效防止鈀的流失和聚集,從而提高其穩定性。隨后,他 們 又 將RuCl3·H2O 負 載 于KAPs 用 于NH4OAc、DMF 與苯乙酮的環化反應以及重氮二羰基化合物與烯烴的環加成反應[43]。該催化劑同樣表現出很好的穩定性,重復使用7 次催化劑中釕無明顯流失。
2014 年,丁云杰團隊[44]改變a12、b10 和c1 比例制得了用于高碳烯烴氫甲酰化的銠納米顆粒催化劑。該研究指出,基于KAPs 的銠催化劑較基于二氧化硅的催化劑具有更大比表面積和孔體積,銠納米顆粒在KAPs 中的分布更加均勻,具有更高的催化活性。但是該催化劑化學選擇性和區域選擇性差,有大量烷烴和異構烯烴生成,直鏈產物和支鏈產物之比也處在0.5~1的范圍內。
隨后,雙膦配體a13[45]、含S/N雜原子的a14[46]、a15[47]的膦配體以及一些含雜原子的共聚單體[46,48]也被應用到含磷KAPs的制備中。景曉飛團隊[47]發現,改變膦配體單體能影響聚合物負載的金納米顆粒粒徑。基于含吡啶基膦配體(a15)的KAPs中生長的金納米顆粒粒徑在1.9nm 左右,而基于三苯基膦(a12)的粒徑分布則在更大2.8nm。無獨有偶,黃延強團隊[48]也在制備KAPs 負載的銀納米顆粒催化劑時發現,基于三苯基膦(a12)的KAPs中生長的銀納米顆粒粒徑均勻分布在4.1nm左右,而無膦配體單體時銀納米顆粒粒徑在9.8nm左右,且粒徑分布不均勻。由此可見,制備基于KAPs 的金屬納米顆粒催化劑時,能通過改變縮聚單體對納米金屬顆粒粒徑的大小和分布進行調控,從而調控其催化性能。
3.2 金屬-膦配合物間的Friedel-Crafts縮聚
2017年,賴志平團隊[49]將Friedel-Crafts縮聚應用到了金屬-膦配合物的縮聚中來,用鈀配合物a16 一步合成了單位點的PPPd 催化劑(圖9)。該催化劑在偶聯反應(Suzuki-Miyaura 交叉偶聯、鹵代芳烴還原自偶聯、芳烴硼酸氧化自偶聯)和C—H 鍵官能團化(烷氧基化、鹵化、烯基化)反應中表現出良好的催化活性和穩定性。2018 年,該團隊[50]又使用釕配合物a17 一步合成用于甲酸制氫的釕催化劑,并通過EXAFS 分析證明了該催化劑釕主要以單原子的形式存在。該催化劑在DMSO、H2O/DMSO 溶劑體系下循環使用50 次甲酸轉化率未見明顯下降,表現出很好的穩定性。值得指出的是,該團隊此前已經證明配合物a17是甲酸制氫的很好的均相催化劑[51]。這再次體現出均相對聚合物基催化劑的指導作用。2019 年,Iglesias 團隊[52]將該方法應用到更多的金屬配合物,如a18、a19 和a20,制備了一系列含釕、金的催化劑。直接用金屬配合物通過Friedel-Crafts縮聚合能合成高金屬/磷比例的金屬催化劑,相較金屬后負載的方式其操作也更加簡便,控制得當還能制備單原子催化劑。

圖9 一步法合成單位點鈀催化劑PPPd
一方面,Friedel-Crafts 縮聚無需使用官能基化的單體而選擇范圍廣,能通過調控單體組分(膦配體單體、非膦共聚單體和偶聯劑及其配比)控制含磷聚合物比表面積和孔徑分布等性質;另一方面,Friedel-Crafts 縮聚合成步驟簡單,既能先合成含磷聚合物再通過后負載制備金屬催化劑,也能直接用金屬配合物一步合成,在含磷多孔有機聚合物基催化劑的制備上具有一定的普適性。但是,Friedel-Crafts 縮聚需要使用FeCl3或AlCl3做催化劑,如果除不凈可能對反應帶來不利影響。此外,通過該法調控膦配體單體改變金屬空間位阻進而控制催化劑區域選擇性的報道還沒有,還需更系統的研究。
本部分提到的單體、聚合物物理性質以及各聚合物基催化劑的應用匯總見圖10和表3。

圖10 Friedel-Crafts縮聚單體
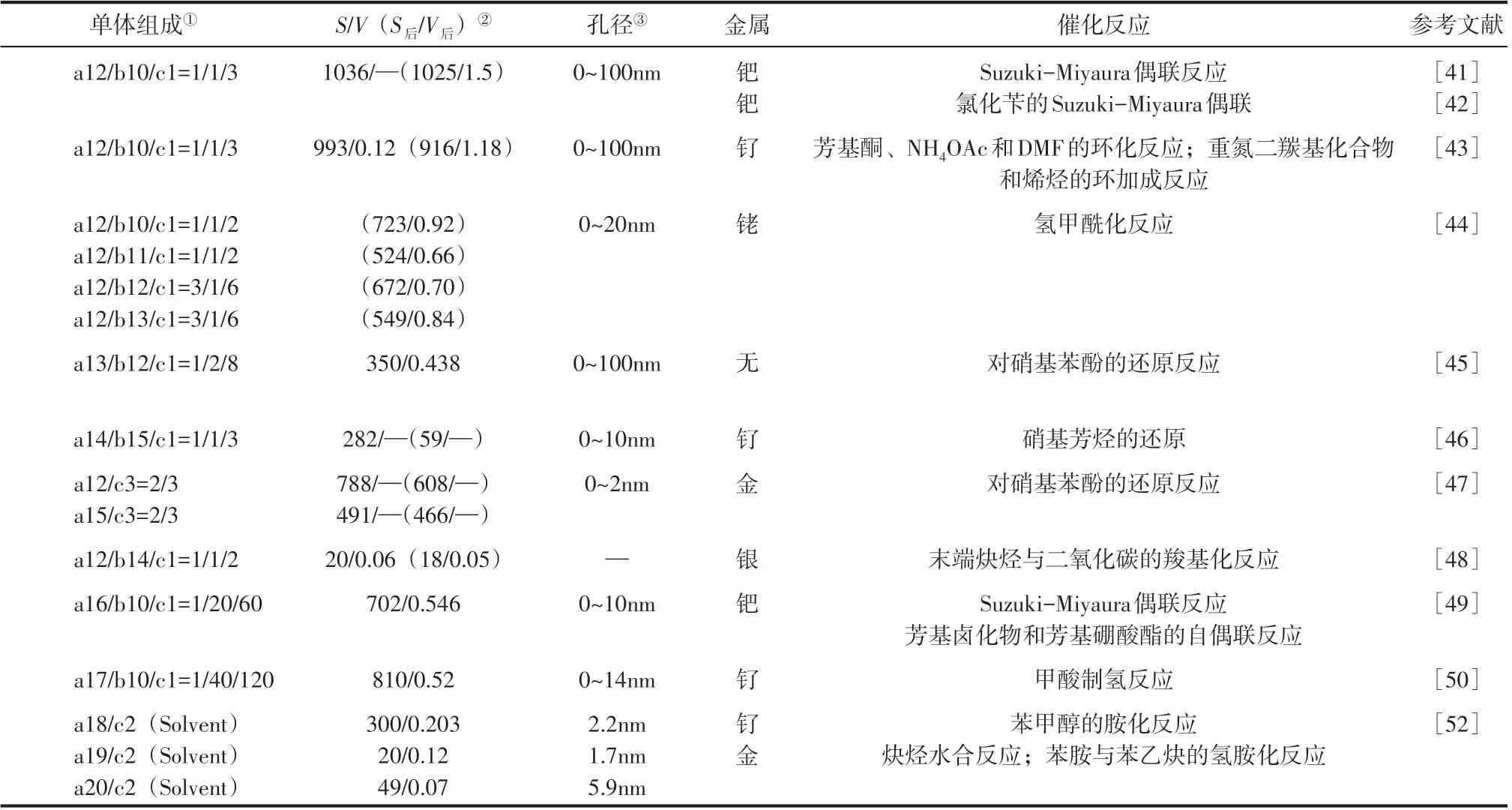
表3 Friedel-Crafts縮聚含磷聚合物單體組成、孔結構參數及其在多相催化中的應用
4 溶劑熱烯烴聚合
早期,通過烯烴自由基聚合制備含磷聚合物載體以可溶的線性聚苯乙烯為代表[10]。隨后,科研人員又利用多烯烴反應位點的膦配體單體通過懸浮聚合[53]或乳液聚合[54]等途徑合成一些交聯的孔基含磷聚合物載體。一般說來,這類聚合物的比表面積較小,孔結構不發達。2009年,肖豐收團隊[55]首次報道了溶劑熱聚合的方法。溶劑熱烯烴聚合,選用適當的溶劑能合成具有發達孔道結構和高比表面積的聚合物。多孔性的含磷聚合物十分適合用作催化劑載體,因此該法在含磷多孔有機聚合物基的催化劑制備中被廣泛應用。迄今為止,被報道的用于溶劑熱烯烴聚合的烯基化的含磷單體已經有數十種,包括烯基化的單膦配體、雙膦配體和季鹽單體。本文總結了通過溶劑熱方式制備的多孔聚合物,其單體組分、物理性質(比表面積、孔體積和孔徑)、負載金屬類型以及相應催化劑的應用,在相應部分的圖表列出。
4.1 單膦配體聚合物
銠與膦配體(三價膦配體、亞磷酸酯配體和亞磷酰胺配體)的配合物是一類活性很高的烯烴氫甲酰化催化劑,例如已經工業應用的HRh(CO)(PPh3)2催化劑,它能在低一氧化碳壓力條件下催化氫甲酰化反應高效進行[56]。2014—2015年,肖豐收團隊和丁云杰團隊[57-58]制備了一系列用于氫甲酰化反應的含磷聚合物基銠催化劑,并對聚合物基催化劑體系進行了系統的研究。該作者用膦配體單體3V-PPh3(a21),通過溶劑熱聚合成功制得比表面積和孔體積分別高達1086m2/g 和1.70cm3/g 的含磷聚合物(POL-PPh3),負載Rh(CO)2(acac)和HRh(CO)(PPh3)3后分別獲得了單位點銠的氫甲酰化催化劑Rh(CO)2(acac)/POL-PPh3和RhH(CO)(PPh3)3/POL-PPh3。兩種聚合物基催化劑表現出與均相催化劑[HRh(CO)(PPh3)3]類似的活性和選擇性,且在釜式反應器和固定床反應器中均能使用。其中,Rh(CO)2(acac)/POL-PPh3于固定床反應器催化乙烯的氫甲酰化 反 應, TOF (turnover frequencies) 值 能 達10534h-1[57]。根據Rh(CO)2(acac)/POL-PPh3在混合氣體(C2H4∶CO∶H2=1∶1∶1)中的原位紅外分析結果,再結合其與HRh(CO)(PPh3)3表現出類似活性和選擇的實驗結果,提出了銠在聚合物中與三個磷原子配位的模型(圖11)。隨后,丁云杰團隊[59]用原位加入和后負載銠配合物兩種方式制備催化劑,發現原位引入方式可能使銠位點被聚合物覆蓋而活性較低。接著,肖豐收團隊[60]探究了聚合物中磷的濃度以及銠/磷比例對催化活性的影響。實驗結果表明高磷濃度的聚合物載體有利于催化劑活性的提高,而銠/磷比例過高或過低都會給催化劑帶來不利影響。

圖11 Rh/POL-PPh3中銠、磷之間的三角錐和平面配位模型
2015—2017年,丁云杰團隊又將PdCl2(PhCN)2[61]、RuCl3[62]和Rh2(CO)4Cl2[63]分別負載于POL-PPh3上應用于催化Suzuki-Miyaura 偶聯反應、肉桂醛加氫反應和甲醇羰化反應。2018 年,雷以柱團隊[64]將Pd(OAc)2負載于POL-PPh3上用于催化苯硼酸化合物或醇類化合物與鹵代芳烴的羰化偶聯。
含磷聚合物載體通過單體設計和共聚的方法賦予其特殊性質,使得聚合物基催化劑具有很好的可調變性。丁云杰團隊[65]通過用a21 與b18~b20 共聚合成一系列用于加速環氧化合物與二氧化碳生成環碳酸酯的催化劑。在甲酯基化反應中,吡啶基取代的膦配體能加速醇的脫質子從而加速酯的生成[66]。基于此,丁云杰團隊[67]合成了基于單體a22的含氮、磷的多孔聚合物,負載Pd(OAc)2后用于炔烴的甲酯基化反應。之后他們使用a21 或a22 與b16 共聚引入苯磺酸基團獲得了多功能甲酯基化反應催化劑[68]和嗎啉氫甲酰化反應(二氧化碳為羰源)催化劑[69]。近來,他們又通過篩選大量基于不同烯基化膦配體單體(a23~a33)的含磷多孔聚合物,開發出了一種用于末端炔烴雙錫基化的聚合物基單原子催化劑[70]。該單原子催化劑聚合物載體基于單體a23,a23 特殊的結構和電子性質使得催化劑具有很高的活性和選擇性。值得注意的是,該作者還指出鈀前體對催化劑中金屬的分散和催化劑結構均有影響,Pd(PPh3)2Cl2的18 電子結構對鈀的單位點分散至關重要。
對于一些水相用催化劑或基于亞磷酸酯配體的聚合物催化劑,其親疏水性會對催化劑的活性或穩定性產生影響[71-73]。雷以柱等[72]通過改變共聚單體a21 與b17 的比例調控聚合物的親水性,實現了對聚合物基催化劑在水相中催化Suzuki-Miyaura 偶聯反應活性的調控。肖豐收團隊[71,73]用亞磷酸酯配體單 體phosphite-tBu (a34) 制 備 了 多 孔 聚 合 物Phosphite-POP,其多孔結構賦予其超強的疏水性(與水的接觸角達到152°)。Phosphite-POP 基銠催化劑相較于均相催化體系Rh/phosphite-tBu 具有更高的活性和穩定性。基于a35 和a36 的多孔聚合物催化劑也存在類似的規律,較其相應的均相催化體系活性高。
除了鈀和銠,其他金屬化合物如AuCl[74]和[Cp*IrCl2]2[75]也被用于與基于3V-PPh3(a21)的聚合物合成多相催化劑。
本部分單體結構、組分、物理性質(比表面積、孔體積和孔徑)、負載金屬類型以及相應催化劑的應用參見圖12、表4。

圖12 用于烯烴聚合的單膦配體單體及共聚單體

表4 溶劑熱合成的含磷聚合物單體物組成、孔結構參數及其在多相催化中的應用(Ⅰ)
4.2 雙膦配體聚合物
基于雙膦配體能獲得高化學選擇性和立體選擇性催化體系[76-77],科研人員設計了一系列烯基取代的雙膦配體單體,用于合成多孔聚合物載體并運用于多相催化。
2012 年,肖豐收團隊[78]改性(R)-BINAP 合成了烯基化的單體a37,經溶劑熱聚合、HSiCl3/PPh3還原成功合成了聚合物PCP-BINAP。該聚合物負載[RuCl2(benzene)]2后應用于β-酮酯的不對稱加氫,選擇性和ee值分別高達99.5%和99.0%。丁云杰團隊[79-80]同樣通過改性BINAP 合成了烯基化單體(S)-4,4'-divinyl-BINAP(a38)和(S)-5,5'-divinyl-BINAP(a39),指出4(4')位乙烯基取代的BINAP(a38)中磷原子受聚合物骨架的位阻作用較5(5')位取代的a39 更大,因此基于a39 的聚合物釕催化劑在β-酮酯的不對稱加氫反應中能達到更高的轉化率和ee值[80]。
2015 年,肖豐收團隊[81]分別用單體a40、a41和a42 通過溶劑熱、后負載Rh(CO)2(acac)的途徑制得了聚合物銠催化劑,并將其用于烯烴氫甲酰化反應。相較a41和a42,基于a40的聚合物骨架靈活性好,其31P 核磁譜圖信號峰在甲苯中由無甲苯溶劑時的寬信號峰轉變成窄而尖的信號峰,表現出“準均相”的性質。該作者認為正是單體a40柔性鏈賦予聚合物載體“準均相”的性質,使得其催化活性高于基于a41 和a42 的聚合物催化劑。當然,這并不是說基于a41 或a42 的聚合物載體的催化劑活性就低,例如在硫代苯甲酰胺和異腈類化合物合成噻唑類化合物的反應中,基于a41的聚合物鈀催化劑就有著很好的活性[82]。隨后,丁云杰團隊報道了一系列用于氫甲酰化反應的其他聚合物基催化劑,這些聚合物載體基于烯基化的雙膦配體單體a39[79]、a43[83-86]和a44[87-88]。其中,基于a43 與a21 共聚物的銠催化劑在C3、C4烯烴的氫甲酰化反應中表現出很高的活性,醛類產物選擇性(化學選擇性)高達94.2%,且醛類產物中正異比(區域選擇性)高達62.2[84-85]。而在長鏈的正辛烯的氫甲酰化反應中雖有著高的區域選擇性,化學選擇性較低(醛類產物占58%左右),相當一部分烯烴轉化成了相應的異構烯烴和烷烴[86]。而基于a44 與a21 的共聚物銠催化劑在正辛烯的氫甲酰化反應中有著良好的活性和穩定性,在高區域選擇性前提下還有著不錯的化學選擇性(醛類產物占87%)[88]。此外,基于a44 自縮聚的鈀納米催化劑能用作脫羰催化劑,適用于苯環醛基、雜環醛基甚至是長鏈醛的脫羰基[87]。2019年,賈肖飛團隊[89]合成了烯基取代的雙亞磷酰胺配體a45,并用于制備烯烴氫甲酰化的聚合物基多相銠催化劑。該催化劑在正己烯的氫甲酰化中表現出超高的活性(TON 達45.3×104),對醛類化合物的選擇性為90.3%且正異比高達49.1。當底物換成正辛烯時,活性略有下降(TON仍有1.6×104),但對醛的選擇性達91.3%且正異比為41。同年,肖豐收團隊[90]合成了超分子離子對膦配體單體a46 和a47,經溶劑熱聚合、負載Rh(CO)2(acac)制成催化劑。該類催化劑能在水溶液中催化正辛烯的氫酯基化反應的進行,轉化率達94.7%~96.3%,醛的選擇性高達98.0%~98.4%且正異比高達39。2020 年,楊勇團隊[91]又合成了新聚合單體a45',基于該單體的聚合物基銠催化劑能用于高碳烯烴的氫甲酰化,產物中醛的正異比高達175。值得注意的是,雙膦配體單體常與單膦配體a21共聚,進而負載銠金屬用于氫甲酰化反應。其中,雙膦配體的螯合作用能提供大位阻環境,而單膦配體能起到穩定銠金屬物種的作用。
上述基于a42、a43、a44、a45或a45'的聚合物銠催化劑在氫甲酰化中表現出高活性和高選擇性,而其膦配體單體對應的雙膦配體在均相體系中早已被證明是具有高活性和選擇性的配體[92]。這是均相催化體系對基于聚合物基催化劑有指導作用的又一有力證明。因此在均相催化劑被大量研究的領域(如烷氧羰基化反應、氫甲酰化反應、不對稱加氫反應等),均相催化體系能為聚合物基催化劑的開發提供很好的指導。
本部分單體結構、組分、物理性質(比表面積、孔體積和孔徑)、負載金屬類型以及相應催化劑的應用參見圖13、表5。
4.3 季鹽聚合物
本部分單體結構、組分、物理性質(比表面積、孔體積和孔徑)、負載金屬類型以及相應催化劑的應用參見圖14、表6。

圖14 用于烯烴聚合的季鹽單體及共聚單體
肖豐收團隊在合成多孔聚合物方面做了諸多開創性的工作,如率先報道了溶劑熱的方法合成多孔有機聚合物[55],最先報道多孔季鹽型聚合物并將其用于多相催化[97]等。丁云杰團隊則拓展了含磷多孔有機聚合物基催化劑在多相催化領域的應用,實現了多相催化氫甲酰化的工業應用[20]。在他們的推動下,溶劑熱烯烴聚合近十年發展發展迅速,不管
是含磷單體還是不含磷的共聚單體都逐漸變得豐富起來。烯烴聚合能在均相催化體系的指導下設計烯基化的聚合物單體,而且也能引入具有特定功能基團的共聚單體制備多功能的催化劑,在結構調控方面具有巨大的發揮空間。此外,共聚組分(單體及配比)、金屬負載量、金屬前體以及負載方式(原位加入、后負載等)等都對聚合基催化劑有影響,這一方面使得聚合物基催化劑的可調變性進一步變大,同時也使得聚合物基催化劑的制備更加復雜。作為聚合物基催化劑中起決定性作用的膦配體單體,其合成路徑通常比較繁瑣且合成條件苛刻,這是溶劑熱法聚合物在多相催化領域走向應用的障礙。

表5 溶劑熱合成的含磷聚合物單體物組成、孔結構參數及其在多相催化中的應用(Ⅱ)

表6 溶劑熱合成的含磷聚合物單體物組成、孔結構參數及其在多相催化中的應用(Ⅲ)
5 其他
其他合成含磷多孔有機聚合物的方法還包括醛胺縮聚、Scholl 縮聚、酚醛聚合、聚吡喃鹽的磷代以及多段式聚合等。
5.1 醛胺縮聚
用多元胺和多元醛化合物通過醛胺縮聚的方式是構建COFs 的常用方法[99]。相比其他含磷多孔有機聚合物,長程有序的骨架結構是含磷COFs 獨有的特點。這種長程有序包括有序分布的膦配體,特定結構且均勻分布的孔道結構。2019 年,張偉團隊[100]使用三(4-甲酰基苯基)膦a56 和對苯二胺b25成功合成了Phos-COF-1(見圖15),其孔徑主要分布在1.56nm 左右,比表面積為818m2/g。Phos-COF-1 與K2PdCl4、K2PtCl4或HAuCl4在甲醇中混合24h,滴加NaBH4還原后能可控合成平均粒徑分別為1.62nm、2.06nm 和1.78nm 的鈀、鉑和金超細納米顆粒催化劑。該作者還用K2PdCl4和HAuCl4的混合金屬配合物溶液制得基于Phos-COF-1 的鉑鈀納米顆粒催化劑,指出該方法的可控性得益于膦配體的配位誘導生長和孔結構的空間限制機制。膦配體與鈀、鉑或金的前體配位,使得金屬離子在Phos-COF-1 均勻分布。金屬離子被NaBH4還原成原子后聚集成納米顆粒,納米顆粒的生長又受到Phos-COF-1 超細孔結構的限制,最終形成粒徑較均勻的超細納米顆粒。此外,Phos-COF-1 超細孔結構還能在催化劑使用過程中有效限制中金屬納米粒子進一步團聚長大,因此具有較好的重復使用性,重復使用5次金屬顆粒只是略有增大。
隨后,范以寧團隊[101]通過類似的方法獲得了P-COF-1和P-COF-2兩種含磷COFs。P-COF-1和P-COF-2分別由二元胺單體b25、b26和三(4-甲酰基苯基)膦a56縮聚而來,負載Rh(CO)2(acac)后用于催化苯乙烯的氫甲酰化反應。Rh-P-COF-1和Rh-P-COF-2 首 次 使 用TOF 值 分 別 達2557h-1和2074h-1,重復使用6 次還能維持在2398h-1和2050h-1,具有較好的穩定性。但醛類產物中直鏈醛與支鏈醛之比僅為1,區域選擇性有待提高。
5.2 Scholl縮聚
Scholl縮聚是芳烴類化合物在酸(AlCl3和質子酸)催化下偶聯縮聚合成聚合物的方法。2014年,譚必恩和李濤團隊[102]首次將Scholl縮聚應用于含磷微孔聚合物的合成,用三苯基膦a12 和芳烴b13 在AlCl3、質子酸的催化縮聚下合成了肖爾偶聯微孔聚 合 物(Scholl-coupling microporous polymers,SMPs),記為SMP-8a(圖16)。SMP-8a 負載PdCl2后制成含鈀催化劑SMP-8b,在Suzuki-Miyaura 偶聯反應中表現出很高的催化活性。Scholl 縮聚合成SMPs簡單方便,不過路易斯酸AlCl3的使用增加了后處理難度。SMPs 通過苯環間的共價鍵直接交聯有利于微孔的形成,其固有的大π共軛結構骨一方面賦予聚合物很強的剛性,致使其溶脹性能較差,另一方面也使得其具有發光和半導體性質。

圖15 醛胺縮聚合成含磷COFs
5.3 酚醛聚合
2018 年,王忠剛團隊[103]通過溶劑熱法成功合成了多孔含磷酚醛樹脂,將三(4-甲酰基苯基)膦(a56)和2,5-二羥基-1,4-苯醌(b27)溶解在二烷中,90℃加熱攪拌1h 制得均勻透明液體,然后將該液體轉移至帶有聚四氟乙烯內襯的水熱釜中,于220℃條件下反應4d制得聚合物體,經洗滌干燥得到含磷酚醛樹脂PFN-P(圖17)。溶劑熱的合成方法使PFN-P 具有很好的多孔性,含有大量的微孔和介孔,比表面積和孔體積分別達775m2/g 和0.72cm3/g。PFN-P 與Pd(PPh3)4的甲苯溶液混合24h得到Pd@PFN-P 催化劑,在Suzuki-Miyaura 偶聯反應中表現出很好的活性和穩定性。
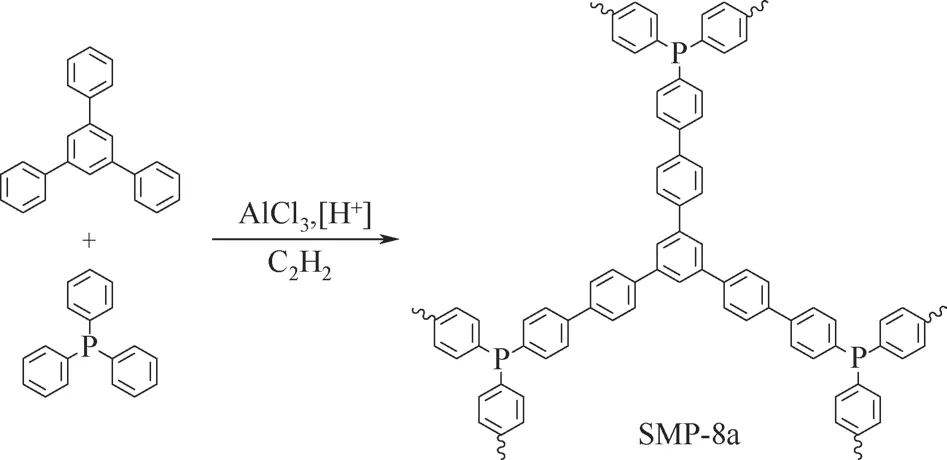
圖16 Scholl縮聚合成含磷微孔聚合物
上述方法合成的聚合物的單體組成、孔結構參數及其在多相催化中的應用見圖18和表7。

圖17 溶劑熱酚醛聚合合成含磷酚醛樹脂

圖18 醛胺縮聚及酚醛聚合單體
5.4 聚吡喃鹽的磷代
磷雜苯為具有平面構型的芳香性磷化合物,理論計算表明其芳香性為苯環的88%[104]。由于其特殊的空間構型和電子性質,磷雜苯與金屬的配位和位阻性質與傳統的三價有機膦配體有很大的不同(圖19)[105]。因此,磷雜苯聚合物在多相催化中可能具有一些獨特的性能。但是磷雜苯很容易被堿[31]或親核試劑進攻[105]發生化學反應,由磷雜苯單體出發直接合成聚合物的是很困難的。

表7 醛胺縮聚、Scholl縮聚及酚醛聚含磷聚合物單體組成、孔結構參數及其在多相催化中的應用
2019 年,戴勝團隊[106]采用“后合成”的策略成功合成了磷雜苯型多孔聚合物(圖20),用單體b28、b29 或b30 分別與四(4-乙酰苯基)甲烷縮聚合成聚吡喃鹽,再經P(Me3Si)3磷化處理成功制得了磷(Pd@HBPs。 HBPs: honeycomb-like bicontinuous P‐doped porous polymers)[108]、超交聯微孔有機納米棒聚合物基金屬納米顆粒催化劑(M-HMONFs。HMONFs: hypercrosslinked microporous organic nanotube frameworks)[109]、中空多孔有機納米球基鈀催化劑[H-PONs-Pd(PPh3)4。H-PONs: hollow porous organic nanospheres][110]。他們首先通過可逆加成-斷裂鏈轉移聚合(reversible addition-fragmentation chain transfer copolymerization,RAFT)合成特定的雜苯型聚合物Phos-POP、F-Phos-POP-1 和FPhos-POP-2。 Phos-POP、 F-Phos-POP-1 和FPhos-POP-2 的比表面積分別為282m2/g、591m2/g和432m2/g,孔徑主要分布在0~2nm的范圍內。此類聚合物負載RuCl3后用作嗎啉的氫甲酰化反應(二氧化碳為羰源)催化劑,結果表明含高濃度氟元素的Ru/F-Phos-POP-2 催化劑表現出相對較高的活性。該作者認為,由于氟對二氧化碳具有親和性,在催化過程中起到了協同作用從而在三種催化劑中表現出最高的活性。雖然磷雜苯型聚合物豐富了含磷配體的電子性質和配位性質,但磷雜苯結構本身不太穩定,易受親核試劑、堿等的進攻。這種變化不僅可能發生在磷雜苯聚合物的合成過程中,也可能發生在催化劑的使用過程中。

圖19 磷雜苯的位阻性質和配位性質

圖20 聚吡喃鹽的合成及磷代
5.5 多段式聚合
通過多種聚合手段的組合能設計合成一些具有特殊形貌結構的含磷多孔有機聚合物,可能在多相催化中表現出特殊的性能。黃琨團隊通過多種聚合手段,控制合成了一系列基于特殊形貌的含磷多孔有機聚合物催化劑(圖21),如微孔有機納米棒聚合物基鈀納米顆粒催化劑(MONFs-PPh3@Pd。
MONFs: microporous organic nanotube frameworks)[107]、蜂窩狀磷摻雜多孔聚合物基鈀納米顆粒催化劑接枝共聚物或嵌段共聚合物;再通過Friedel-Crafts縮聚使含苯環部分交聯成形,將聚酯鏈段刻蝕制成特定形貌的含磷多孔聚合物材料;最后,將金屬前體負載于聚合物材料上制得負載型金屬催化劑。相較本文中提到的其他方法,多段式聚合法通過對形貌的可控構建增加聚合物催化劑的多孔性,尤其是介孔和大孔。介孔和大孔結構有利于傳質,使底物在催化劑體相中能更快的向活性位點擴散。

圖21 基于特殊形貌的含磷多孔有機聚合物催化劑
6 結語
不同含磷多孔有機聚合物的制備方法中選用的單體類型是不同的,縮聚反應的難易程度也各不相同,這使得合成的聚合物在骨架結構、溶脹性、規整度、表面性質(比表面積、孔徑及孔基分布等)等性質各不相同。偶合縮聚制得的聚合物一般孔徑分布較窄,多含微孔或孔徑偏小的介孔。鋰鹽與氯化磷的縮聚反應很容易進行且不可逆,結構無序度高,孔徑分布較寬,制得的聚合物多為含微孔、介孔和大孔的多級孔材料。Friedel-Crafts縮聚發生在偶聯劑與芳香環之間,苯環上反應位點的不確定性使得聚合物孔結構呈無序分布。溶劑熱烯烴聚合為自由基反應,聚合物結構包含大量的柔性烷基鏈段,通常能表現出很好的溶脹性能。醛胺縮合反應具有一定的可逆性,能合成結構長程有序的COFs。Scholl 縮聚合成的聚合物通過苯環間碳碳鍵相連,剛性大且主要含微孔結構。磷化聚吡喃鹽的方式,通過“后合成”的策略能合成不穩定的磷雜苯類聚合物。多段式的聚合策略,能合成特殊形貌結構的聚合物載體。除了制備方法,縮聚組分(縮聚單體及單體配比)、縮聚催化劑的量、后處理方式等因素對聚合物的物理性質(比表面積、孔結構等)也有影響。此外,這些合成方法中偶合縮聚、Friedel-Crafts 縮聚和Scholl 縮聚等需要金屬催化劑參與;溶劑熱烯烴聚合、醛胺縮合和酚醛聚合等無需金屬催化劑參與;Friedel-Crafts 縮聚和Scholl 縮聚所用單體不需要在膦配體單體引入特定反應位點。每種方法都有其優點和缺點,它們互為補充,豐富了含磷多孔有機聚合物單體類型和結構性質。
穩定性是多相催化劑的一項關鍵指標,對于聚合物基催化劑來說,影響其穩定性的有多種因素:金屬于聚合物中的分布、膦配體濃度、聚合物骨架性質等。金屬分布于微孔結構中聚合物骨架對其遷移的阻礙能力比介孔和大孔強,減緩金屬間的團聚以及金屬的流失。聚合物中磷的濃度越高,其對金屬穩定效果越好,對催化劑的重復使用性有一定幫助。在水敏感的聚亞磷酸酯基催化劑中,聚合物骨架強的疏水性對催化劑穩定也能起到關鍵作用。
金屬前體能影響金屬在聚合物中的分散形式。總體來講,以零價金屬配合物為前體有利于形成金屬納米粒,而以相對穩定的金屬離子的配合物(滿足18 電子規則的金屬配合物)為前體則有利于金屬的原子級分散。對于聚合物基金屬納米粒子催化劑,聚合物中膦配體對納米粒子在聚合物中的分散度、生長、催化性能均有影響。膦配體能誘導金屬前體在聚合物載體中均勻分布,在還原團聚過程中能調控金屬顆粒的生長,最后納米顆粒表面電子性質也受其調控。此外,聚合物孔道也能限制金屬納米顆粒的生長,調控納米粒子粒徑。聚合物基金屬單原子或單位點催化劑中,膦配體不僅能調控金屬電子性質影響催化劑活性,還能調控其空間位阻控制催化劑的化學選擇性和區域選擇性,表現出與均相催化體系中膦配體相似的作用。文中有大量的例子說明,在均相催化體系中具有很好活性或選擇性的膦配體,其類似結構被引入聚合物載體后能表現出與均相體系相當甚至更好的活性和選擇性。均相催化劑體系對聚合物基催化劑體系具有指導意義,比傳統無機載體催化劑能更容易得到高活性的催化劑。
前文也提到,大部分合成含磷多孔聚合物的方法都要使用到特定官能基化的膦配體,而官能團化含磷單體的開發和大量合成正是含磷聚合物基催化劑走向應用的一大挑戰。還有一點需要指出,本文提到的含磷聚合物基催化劑絕大部分以負載貴金屬為主,研究非貴金屬催化劑方面的工作很是缺乏。加強對含磷聚合物基非貴金屬催化劑的研究與開發,對降低催化劑制備成本、推進工業應用進程具有重要意義。雖然科研人員發現聚合物能對金屬納米顆粒或金屬單原子的電子、空間位阻等方面產生影響,但由于缺少研究兩者間相互作用的工具,科研人員對聚合物與金屬之間作用的理解還很有限。而研究這種關系至少存在兩大難點:一是影響含磷聚合物結構的因素多,使得對其可控合成變得十分復雜;二是聚合物催化劑中金屬與聚合物配體間的作用是個變化的動態過程。因此還需引入更多動態、原位表征手段或者建立計算模型,以幫助研究人員更好地理解這種作用,進而開發出更高效、穩定的催化劑。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
