Cornelia de Lange綜合征1例新發基因突變報道并文獻綜述
2021-03-11 03:21:02李文靜閆有圣童笑梅
中國生育健康雜志 2021年2期
關鍵詞:特征
李文靜 閆有圣 童笑梅
Cornelia de Lange綜合征(CdLS)是一種較為罕見的遺傳性疾病,1933年荷蘭的兒科醫生Cornelia de Lange對2例有相同臨床特征的患兒進行了首次歸納總結,并以其名命名該類疾病[1]。隨著對該疾病認識的不斷深入,上世紀90年代,Ireland等[1]依據患者的臨床特征,將該疾病分類為典型和輕型兩類。進入本世紀以后,Kline等[2]系統地制定了CdLS的診斷標準,該診斷標準囊括了全球范圍內出現的各種表型,可以準確的對患兒進行分類。
迄今為止,全球范圍內都有CdLS發病報道,該類疾病的發生無明顯的種族傾向,這可能與該類疾病發病率低、臨床工作中對于該疾病的認識不充分[3]、存在漏診和漏報、數據的遺失和不充分,導致目前對于其流行趨勢的認識并不準確所致[4]。為深化對該疾病的認識,本文分析總結了本院收治的1例患兒和國內文獻報道的31例CdLS患者的主要臨床特征,對其臨床表型和相關基因突變進行分析,旨在為臨床工作提供一定的指導。
一、病例資料
1.臨床資料:
(1)入院情況。胎齡37+6周女嬰,生后13 min,主因“窒息復蘇后13 min”收入新生兒病房。患兒經陰道娩出,出生體重2 360 g,羊水III度,胎盤、臍帶無異常,生后Apgar評分1 min 6分(呼吸-1,反應-1,肌張力-1,膚色-1),5 min 9分(反應-1),10 min 10分。孕11周超聲示胎兒頸項透明層增寬,孕20周行羊膜穿刺檢查胎兒染色體核型未見異常,孕21周超聲示單臍動脈,胎兒室間隔基部缺損不除外,孕24周查單核苷酸多態性未見異常,孕30周羊水增多。母親妊娠合并糖尿病,患兒父母非近親結婚,否認家族性遺傳病史,家族成員無類似者(見圖1家族譜系圖)。

圖1 家族譜系圖
(2)體格檢查。脈搏140次/分,呼吸45次/分,血壓 78/37 mmHg,頭圍 31 cm(P10),身長42 cm(
(3)實驗室檢查。血氣、血常規、生化、尿便常規、TORCH-IgM均未見異常;胸片示13對肋骨(見圖4);超聲心動圖(生后24 h)示右心增大,動脈導管未閉3 mm,卵圓孔未閉2 mm,肺動脈高壓74 mmHg;聽力篩查雙耳未通過;顱腦超聲示顱內結構未見異常,腦血流異常;心電圖、眼底檢查、腹部超聲、振幅整合腦電圖、先天遺傳代謝病篩查均未見異常。基因檢測數據經生物信息學分析發現,在NIPBL基因(NM_133433.3)上發現了1個可疑致病變異,c.4986_4992dup(見圖5),先證者資料及父母血樣未檢測到該變異,考慮為新發突變。患兒NIPBL基因出現重復突變,導致mRNA閱讀框發生了框移,提前終止,產生變異蛋白(p.Thr1665HisfsTer2),使蛋白質失去了功能。查閱ClinVar、OMIM 和HGMD等數據庫均未見該變異的報道,屬于新突變。根據美國醫學遺傳學與基因組學學會(AGMG)遺傳變異分類標準與指南[5],判定該變異屬于致病變異。
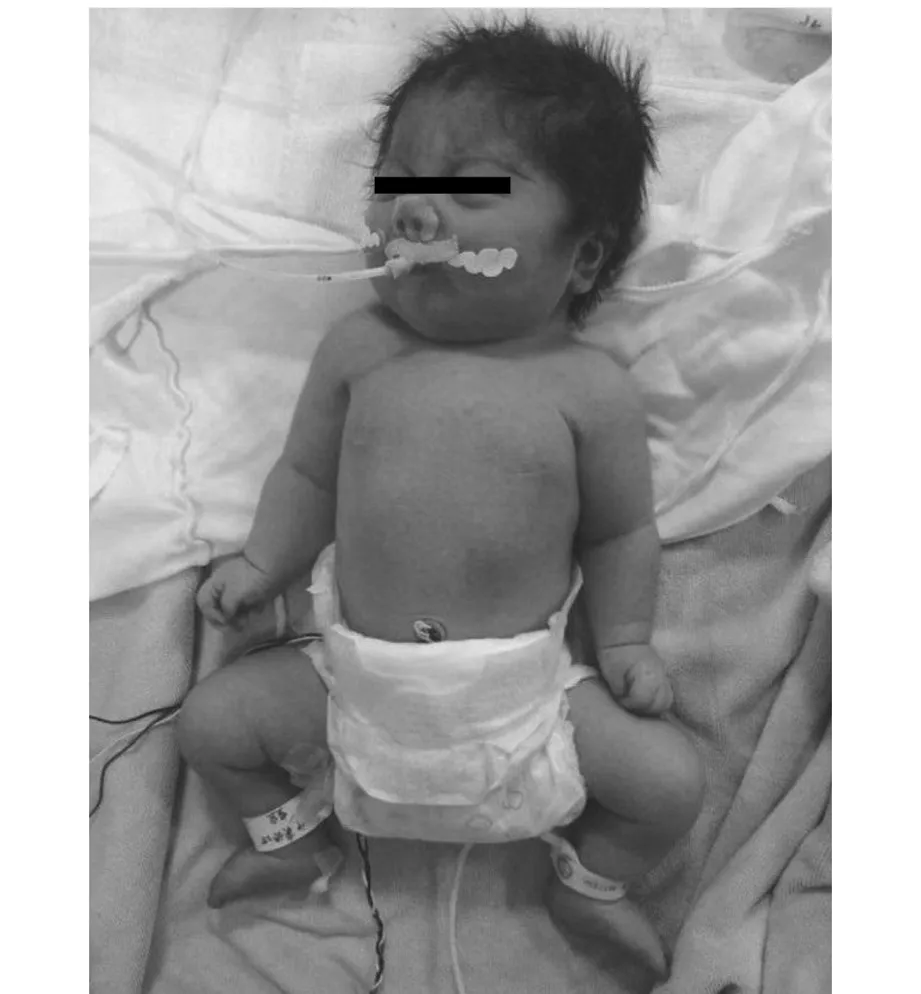
圖2 患兒面容及外觀圖1
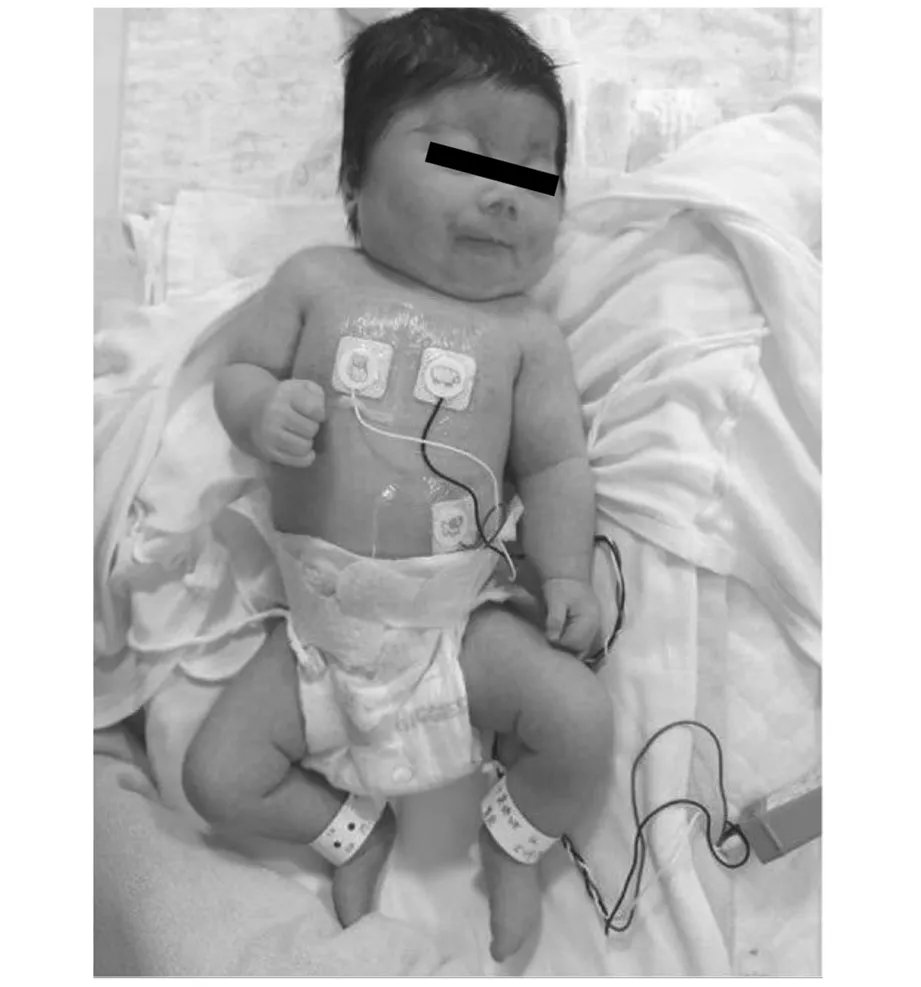
圖3 圖3 患兒面容及外觀圖2
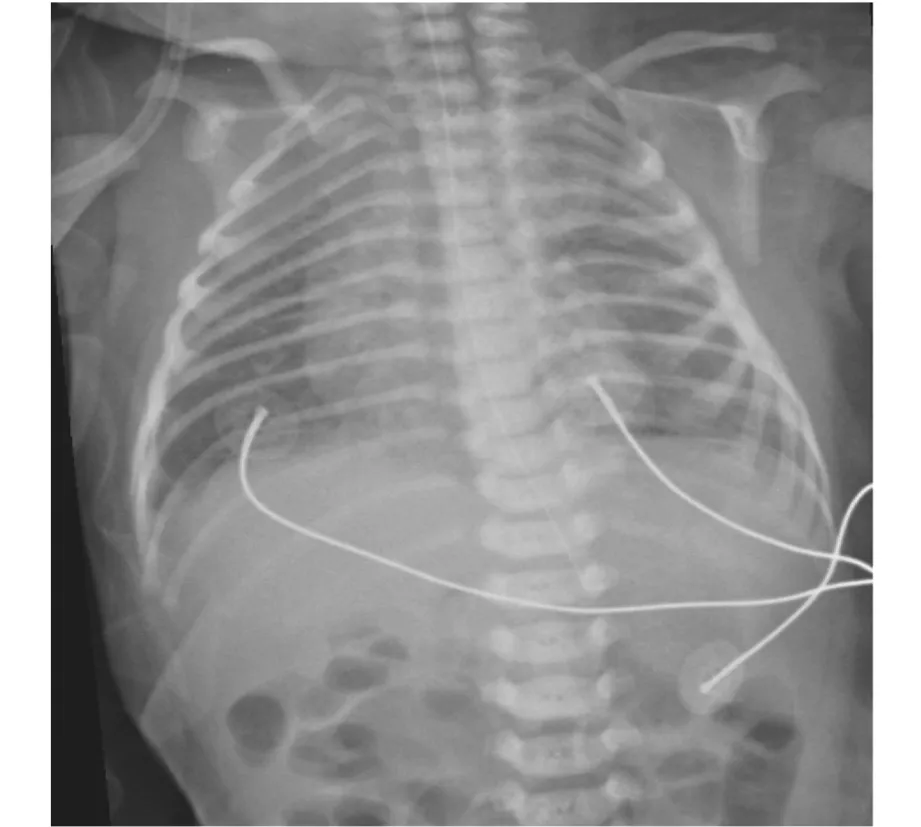
圖4 患兒胸部X線片
(4)診療經過。患兒入院后陣發性呼吸急促,予經鼻高流量吸氧輔助呼吸支持治療,生后6 h呼吸急促逐漸緩解。監測心率、血壓正常,臨床上未出現煩躁、易激惹、驚厥等神經系統異常表現。生后第6 天,家屬要求停止治療接患兒出院。出院2 d后疑因胃食管反流窒息死亡。
2.文獻資料:
檢索1979—2019年該疾病的報道及文獻綜述,檢索數據庫包括PubMed、Web of Science、Medline、中國知網、萬方,發現國內該類疾病的報道共有個案26篇[6-32],碩士畢業論文2篇[33-34],共涉及31例患兒。其中男女比例為11:20。出生胎齡29~40周,其中18例足月,10例早產,3例不詳。平均出生體重2 113 g,其中3例體重不詳。患兒父母均非近親結婚。
國內確診CdLS典型外貌特點有毛發濃密(93.5%)、連眉(90.3%)、指尖細小(82.1%)、頭短小(74.1%)、睫毛長(74%)、通貫掌(67.7%)、口唇薄且角向下(61.3%)、發際低(54.8%)、耳位低(51.6%)、鼻梁扁平朝前(51.6%)、人中淺長(51.6%)、腭裂(6.4%)和雙眼突出(6.4%)。
因文獻中對疾病表現的特征統計項目有差異,患兒全身其他系統異常發生情況存在漏報及缺失,完善相關統計如下(發生例數/統計例數):聽力異常3/3,腦電圖異常3/3,頭顱影像異常3/8,心臟畸形12/23,體重增長緩慢8/8,反復嘔吐以及喂養困難15/16,肋骨數目及形態異常4/4,指骨異常21/31,肌張力異常15/22,智力發育低下21/24,生殖器異常12/24。31例患兒中,24例患兒完善了染色體檢查均未見異常。12例基因檢測中9例發現致病基因,均為NIBPL。
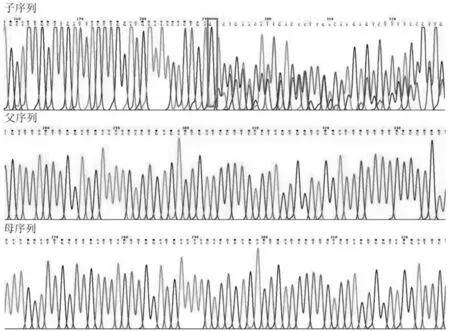
圖5 患兒基因檢測結果圖
二、致病基因及其相關的分子機制
CdLS的發病機制尚未有確定結論,文獻中認為相關的致病基因主要有NIPBL、SMC1A、RAD21、SMC3、組蛋白去乙酰化酶8(HDAC8)、溴結構域蛋白4(BRD4)、錨蛋白重復域11(ANKRD11)[35]。7個主要的致病基因參與構成了Cohesin復合體,這個復合體被稱之為Cohesin通路[36],是一個粘連蛋白復合體,與CdLS的發病密切相關。這個復合體在進化上高度保守,廣泛存在于真核細胞生物,由SMC1A蛋白(structural maintenance of chromosomes 1A)、SMC3蛋白(structural maintenance of chromosomes 3)、STAG蛋白(stromal antigen)、RAD21蛋白(Double-strand break repair protein rad 21 homologue)組成,能夠維持基因的穩定性,廣泛參與DNA的修復和基因的轉錄調節。目前,研究認為基因突變導致了在生長發育過程中粘連蛋白復合體的功能異常,從而影響到一系列下游的基因調控,最終導致疾病發生[37]。
三、CdLS的臨床特點
1.頭面部特征:CdLS患兒頭面部特征最為典型,可以在新生兒時期很容易辨別出,主要表現為低發際線、多毛、連眉、長睫毛、高腭弓、鼻梁低陷、短鼻、鼻孔前傾、長并突出的人中、缺齒、寬齒縫、唇腭裂、口唇薄、口角下垂、低耳位,有報道患兒存在中耳內耳畸形,為中耳炎易發人群,因發育異常,患兒存在傳導性聽力損傷及感音神經性聽力損傷等[35,2]。
2.骨骼及肌肉系統特征:CdLS患兒多有上肢異常,比例約占90%,主要表現為雙手短小、第一掌骨短、小指內彎畸形、貫通掌、橈骨頭脫位、缺指。軀干部多見髖部脫位、脊柱側凸、頸部畸形及漏斗胸,下肢異常較少,可見足部較正常比例偏小,偶見并趾畸形[35,2]。
3.消化系統異常:CdLS患兒胃食管反流情況較多見,反流誤吸為其主要致死因素,其中幽門梗阻導致的消化道梗阻誘發胃食管反流情況最為多見,偶可見環狀胰腺、腸道異常旋轉、梅克爾憩室、肛門閉鎖及先天性膈疝[2]。
4.循環系統異常:部分CdLS患兒存在心血管異常,主要表現為室間隔缺損、肺動脈狹窄及房間隔缺損等[38]。
5.泌尿及生殖系統異常:CdLS患兒主要的泌尿系統畸形包括腎發育不全及膀胱輸尿管返流[39],男性可見隱睪、尿道下裂及陰莖短小,女性可見異常子宮及小陰唇畸形。
6.智力發育障礙及行為異常:該疾病患兒只有3%~4%語言能力接近正常,34.6%存在表達性語言障礙[40]。65.2%的患者出現行為異常,主要的行為異常包括了睡眠障礙、煩躁易怒、注意力不集中等[40-41]。
四、診斷標準及治療
隨著對該疾病認識的逐步深入,2018年10月達成了首部CdLS國際共識[42],該共識將CdLS的表型區分為臨床特點和體征,表型分為了主要特征和提示性特征。主要特征指的是CdLS患者最具典型意義的特征,每一項在評分中積分2分,主要特征包括連眉或者濃眉、鼻梁低陷、鼻短小及鼻孔向前、淺人中或長人中、口唇薄和口角下垂、少指或者無指、先天性膈疝。提示特征指的是這些表現并不夠典型,每項積分1分,提示性特征主要包括宮內發育遲緩、出生后發育遲緩、智力障礙、小頭畸形、小手足、第5指較短、多毛。在表型積分的基礎上,制定了CdLS的臨床診斷標準[41](見表1)。

表1 Cornelia de Lange綜合征臨床診斷標準
國際共識對分子檢測的建議是首選二代基因測序,應包含目前所知的7個主要的致病基因,若條件受限,首選NIPBL基因的Sanger測序。針對非經典表型的人群,可依據臨床經驗選擇性的基因測序,或者進行全基因組或者全外顯子測序。若未檢測到相關的基因突變,建議進一步的完善嵌合變異檢測。
近年來,CdLS的治療尚無突破性進展,當前主流治療方案依然是對癥治療[2,35,42],對于存在顱面部骨性的結構異常,導致上呼吸道發育受限的患兒,建議應盡早手術解除受限情況[42]。De Graaf等[43]報道了1例采用了生長激素治療的CdLS患兒,證實該方案可以有效改善身高發育情況。
五、總結
目前,國內對于CdLS的診斷和治療還存在認識上的欠缺,主要是因為該疾病的發病率低,同時漏診及漏報比例較高,因此針對國內患兒該疾病的特點和表型的歸納和分析的研究都較少。已有學者在進行不同民族和種族本疾病之間差異的研究,2018年Dowsett等[44]比較了4種不同種族的CdLS患兒的面部特點,局限于樣本量,并無特別有意義的陽性發現。因此,完善東亞人群該疾病的研究和性狀統計,對后續相關工作的展開具有一定的指導意義。本例患兒的基因測序提供了新的基因突變證據,能夠為遺傳咨詢和產前診斷提供依據,擴展了突變基因譜,能夠為該疾病的診斷和治療提供一定的指導。
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28
中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16
數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36
空間科學學報(2020年2期)2020-04-01 03:50:40
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
中等數學(2019年8期)2019-11-25 01:38:14
當代陜西(2019年10期)2019-06-03 10:12:04
新聞傳播(2018年11期)2018-08-29 08:15:24
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
