氫氧根交換膜燃料電池陽極氫氧化反應中Ru基電催化劑的進展*
2021-02-15 07:44:38孟凡超王海斌孟祥智胡冠群李春雷叢媛媛趙秋萍
化工科技 2021年6期
關鍵詞:催化劑
孟凡超,王海斌,孟祥智,胡冠群,李春雷,叢媛媛,趙秋萍
(蘭州理工大學 石油化工學院,甘肅 蘭州 730050)
在過去的幾年里,人們對于能源的需求不斷增加,導致了化石燃料的消耗不斷增加,環境污染和氣候變化問題也隨之而來。這些都在迫使人們去探索更加清潔環保的可再生能源。在各種新能源中,由于氫能源有著零污染和能量密度高的特點,使得氫能源備受關注并被認為是最有前途的能源之一。燃料電池可以將化學能直接轉化為電能,且該過程不受卡諾循環的影響,具有清潔和能量轉換效率高等優勢[1]。隨著質子交換膜燃料電池(PEMFCs)技術飛速進步,氫燃料電池汽車一進入市場就大受好評[2]。但是目前的PEMFCs嚴重依賴于Pt基電催化劑,這限制了其商業化發展。氫氧根交換膜燃料電池(HEMFCs),又稱堿性聚合物電解質燃料電池[3](APEFCs),其由PEMFCs衍生而來,使用主鏈上接有陽離子用來傳遞OH-的聚合物電解質膜替代用來傳遞H+的全氟磺酸膜,將酸性環境轉變為堿性環境。就目前而言,HEMFCs也面臨著和PEMFCs同樣的困境,那就是依賴Pt而成本高昂。Pt及其合金仍然是HEMFCs氧還原反應(ORR)和氫氧化反應(HOR)的最佳電催化劑[4]。對于HEMFCs陰極ORR,目前已研發出一些性能優異的非貴金屬電催化劑,如Fe-N-C、Co-N-C等可以替代Pt基ORR電催化劑,大大降低燃料電池陰極側的成本[5]。為了使HEMFCs性能可以與PEMFCs相媲美,需要在陽極側噴涂較高載量的Pt。這是因為當電解質由酸性變為堿性時,Pt的堿性HOR活性降低了近100倍,需提高Pt的載量以加速反應速率[6-7]。陽極側成本的提高削弱了陰極低成本的優勢。因此,開發高效、穩定、低成本的低Pt或無Pt堿性HOR電催化劑對于HEMFCs的發展至關重要[8-9]。
盡管在過去的幾年里陸續報道了非貴金屬的堿性HOR電催化劑,但是其催化活性以及穩定性依然遜色于貴金屬電催化劑[10]。近年來,研究學者們發現Ru具有豐富的氧化還原電化學性質,且價格僅為Pt的6%~36%,存在著明顯的成本優勢,是替代Pt的最佳候選者。純金屬Ru,作為堿性HOR電催化劑,因其本身活性不高且在高的陽極電勢下容易發生氧化[11]。因此圍繞引入其他組分,調變Ru的電子分布,提高Ru基電催化劑的活性,開展了相關工作。
性能優異的Ru基電催化劑的制備依賴于堿性HOR機理的認識。作者闡述了在堿性介質中HOR的電催化機理,說明了影響HOR活性的因素,在此基礎上總結了幾十年來陽極Ru基電催化劑的技術進展并對未來的研究方向提出了展望。
1 堿性介質中HOR的作用機理
在堿性介質中,HOR反應過程可以分為以下3個步驟。

(1)
(2)
(3)
式中,Had表示吸附態的氫,*表示電催化劑的活性位點[12]。
反應速率決速步驟(RDS)是一系列化學反應當中速率最慢的步驟,改善RDS的速率有利于提高整個反應過程的速率[12]。Shinagawa等[13]根據RDS的不同將堿性HOR反應歷程分為Tafel(RDS)-Volmer、Tafel-Volmer(RDS)、Heyrovsky(RDS)-Volmer和Heyrovsky-Volmer(RDS)4類。
對于堿性HOR性能評價而言,存在著2個活性描述符,一個是氫鍵能(HBE),另一個是親氧性(OHBE)。研究學者們對于2個活性描述符在反應中僅氫鍵能起決定作用還是共同作用還沒有達成統一(見圖1)[14]。
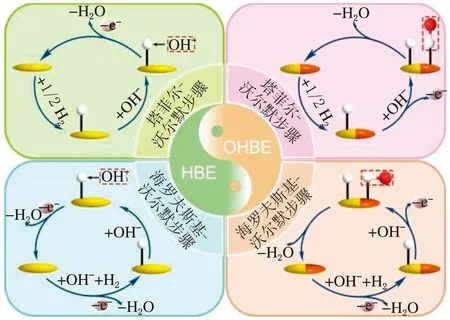
圖1 堿性HOR中HBE和OHBE的理論示意圖
一種觀點認為,反應的活性只與氫鍵能有關。通過Sabatier[15-17]規則可知,電催化劑對H的吸附不能太強也不能太弱,當吸附過強時中間物質的脫附就會變得困難,然而當吸附太弱時,H的覆蓋度就會不足。Sheng等[18]贊同HBE理論,認為該理論與在酸性介質中相同,Had是HOR過程中唯一的中間吸附物質,如果能夠達到最佳的HBE就可以保持Had的吸附和脫附平衡。因此提出可以通過改變條件達到最優的HBE從而實現催化活性的最優化。Yan等[19]發現,Pt的HBE隨著電解質pH值的增加而增加,在堿性條件下,Pt的HBE偏離理想狀況,使得HOR活性降低,故其認為HBE是HOR的唯一活性描述符。Ohyama[20]課題組探究了Ru3Ir2/C和RuIr/C在堿性溶液中的HOR活性,研究結果表明在堿性介質中Ru3Ir2/C的活性要優越于RuIr/C,其主要原因是因為Ru3Ir2/C的合金化使得其HBE相較于RuIr/C降低了。
另一種觀點是HBE和親氧性共同影響堿性HOR活性。Markovic[21]課題組在研究中發現,用Ni(OH)2修飾Pt單晶,其在堿性中的活性會提升大約8倍,將這種活性的提升歸功于親氧組分的引入有利于捕捉含氧物種。Li等[22]在溶液中加入RuCl3時,Pt/C的活性會大大提高,認為通過電位掃描,電催化劑表面引入了親氧性強的組分,提高了電催化劑的堿性HOR活性。此外,Strmcnik等[23]也支持隨著親氧位點的增加,電催化劑的堿性HOR活性也會隨之增加。Duan[24]課題組的研究發現MoNi4相較于純Ni電催化劑,其活性顯著增加,這是因為Ni和Mo的合金化降低了HBE,與此同時引入的Mo增強了親氧性,大大促進了Volmer步驟,提高了堿性HOR活性。Alesker等[25]將Pd和Ni的混合納米顆粒(NPs)用作HEMFC的堿性HOR電催化劑,其峰值功率密度(PPD)為0.40 W/cm2是純Pd(0.18 W/cm2)電催化劑的2.2倍。提出在不考慮合金化和電子效應的影響下,NPs中Ni的親氧性是提升HOR活性的關鍵所在。Liu等[26]通過原位X射線吸收譜(XAS)觀察到了在HOR的電位區間內存在OHad,并進一步提出了OHad通過雙功能機理促進了Volmer步驟進而促進了堿性HOR反應。Ishikawa[27]課題組合成了Ru-Ir/C電催化劑,通過測量其Tafel斜率發現Volmer步驟是HOR的RDS,同時Tafel步驟也在影響著整個反應,因此提出了堿性HOR反應在Ru-Ir電催化劑表面上通過雙功能機理而得以增強。當然也有學者對這個說法提出了異議,稱并非所有的條件下都適用于此機理,比如Ramaswsamy等[28]就提出了在Pt與Nb、Ni、Cu等過渡金屬形成的合金表面,在高pH值條件下不是形成Hupd而是一層氧化物,此時才涉及到HOR雙功能機理,即Pt-Had與過渡金屬氧化物上形成的OHad直接發生反應。Liu等[29]同樣發現只有約0.9 Vvs相對于標準氫電極(vs. RHE)時,OHad具有更好的穩定性,更可能參與到反應中,這時便遵循雙功能機理。
2 Ru基電催化劑
Ru金屬價格低廉,是目前有潛力替代Pt的陽極電催化劑,吸引了越來越多的學者。然而,Ru的堿性HOR活性一直低于Pt,這是由于Ru具有較強的氫鍵能以及親氧性[11],因此需要引入其他組分,優化電催化劑的氫鍵能或親氧性,以提高電催化劑的活性。
2.1 Ru-M合金電催化劑
通過Ru金屬與其他金屬形成合金作為燃料電池的電催化劑,其堿性HOR電催化劑的活性以及穩定性可以得到顯著提高,因為合金組成以及晶體結構的調變會改變電催化劑中Ru的電子結構,從而優化電催化劑的氫鍵能或親氧性,提高堿性HOR活性。
Papandrew[30]等人基于化學氣相沉積工藝合成了分散在碳載體上的Ru基RuxMy(M=Pt或Pd)合金電催化劑,表征結果顯示決速步驟取決于合金材料的選擇,在合成的一系列合金化合物中Ru0.20Pt0.80和Ru0.20Pd0.80表現出最佳活性,碳擔載的Ru0.20Pt0.80和Ru0.20Pd0.80電催化劑的HOR面積比交換電流密度(j0,s)分別是1.42和0.148 mA/cm2,是Pt/C(0.49 mA/cm2)和Pd/C(0.05 mA/cm2)的3倍。
Ishikawa[27]等人通過多元醇法合成了碳擔載的不同金屬比例的Ru-Ir合金納米顆粒電催化劑(Ru-Ir/C),與Ru/C和Ir/C相比,所有Ru-Ir/C合金電催化劑均表現出更高的活性,這表明合金化可以有效提高堿性HOR活性。當Ru/Ir的原子比為2∶3時,其j0,s大約為0.53 mA/cm2,分別是純Ru和純Ir電催化劑的19.5倍和3.5倍。
Scofield[31]團隊合成了Pt7M3合金納米線(NWs,M=Ru、Fe、Co、Cu、Au),在測試中發現Pt7Ru3NWs表現出最高的HOR活性,在0.05 V vs.RHE條件下表現出了2.2 mA/cm2的活性以及0.493 mA/cm2的面積比活性,優異于純Pt NWs(1.38 mA/cm2和0.229 mA/cm2)。
Cong[32]等人通過濕法浸漬,高溫還原和高溫NH3蝕刻合成了負載在氮摻雜碳(N-C)上的一系列Pt1-xRux顆粒(見圖2)[31],在一系列的合成物中Pt0.25Ru0.75/N-C表現出了最高的堿性氫氧化活性,其質量比交換電流密度(j0,m)為1 654 A/g PtRu,分別是商業Pt/C(352 A/gPt)和PtRu/C(1 213 A/g PtRu)的4.7倍和1.4倍。
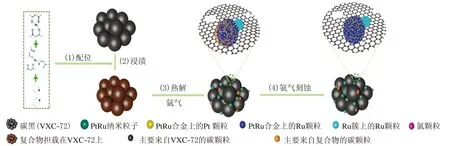
圖2 Pt0.25Ru0.75/N-C電催化劑的合成示意圖
Wang[33]的課題組為了研究合金化對于催化活性的影響,合成了Ru-M/C(M=Co、Ni、Fe)電催化劑,在實驗過程中發現隨著Co、Ni或Fe含量的增加,電催化劑H的吸附/解離過程的可逆性增強使得HOR活性也隨之增強。其所有的Ru0.95M0.05/C(M=Co、Ni、Fe)電催化劑的活性均高于Ru/C,有的甚至高于Pt/C。Ru0.95Co0.05/C的活性要略高于Ru0.95Ni0.05/C和Ru0.95Fe0.05/C,其j0,m是純Ru的5倍。
Xue[34]等人報道了一種無鉑電催化劑Ru7Ni3/C,與先前報道的Ru基電催化劑相比,其具有優異的堿性HOR活性,在0.05 V vs.RHE處的質量比活性為9.4 A/mgRu,分別為Ru/C(0.28 A/mgRu)、Pt/C(0.45 A/mgPt)和PtRu/C(3.5 A/mgPt+Ru)的33、21和3倍。在0.05 V vs.RHE處的面積比活性為23.4 mA/cm2,分別是Ru/C(0.67 mA/cm2)、Pt/C(0.93 mA/cm2)和PtRu/C(4.8 mA/cm2)的35、25和5倍(見圖3)[34]。如此優異的性能要歸功于Ni與Ru的合金化作用以及表面氧化的Ni物種共同削弱了Ru-H鍵強。
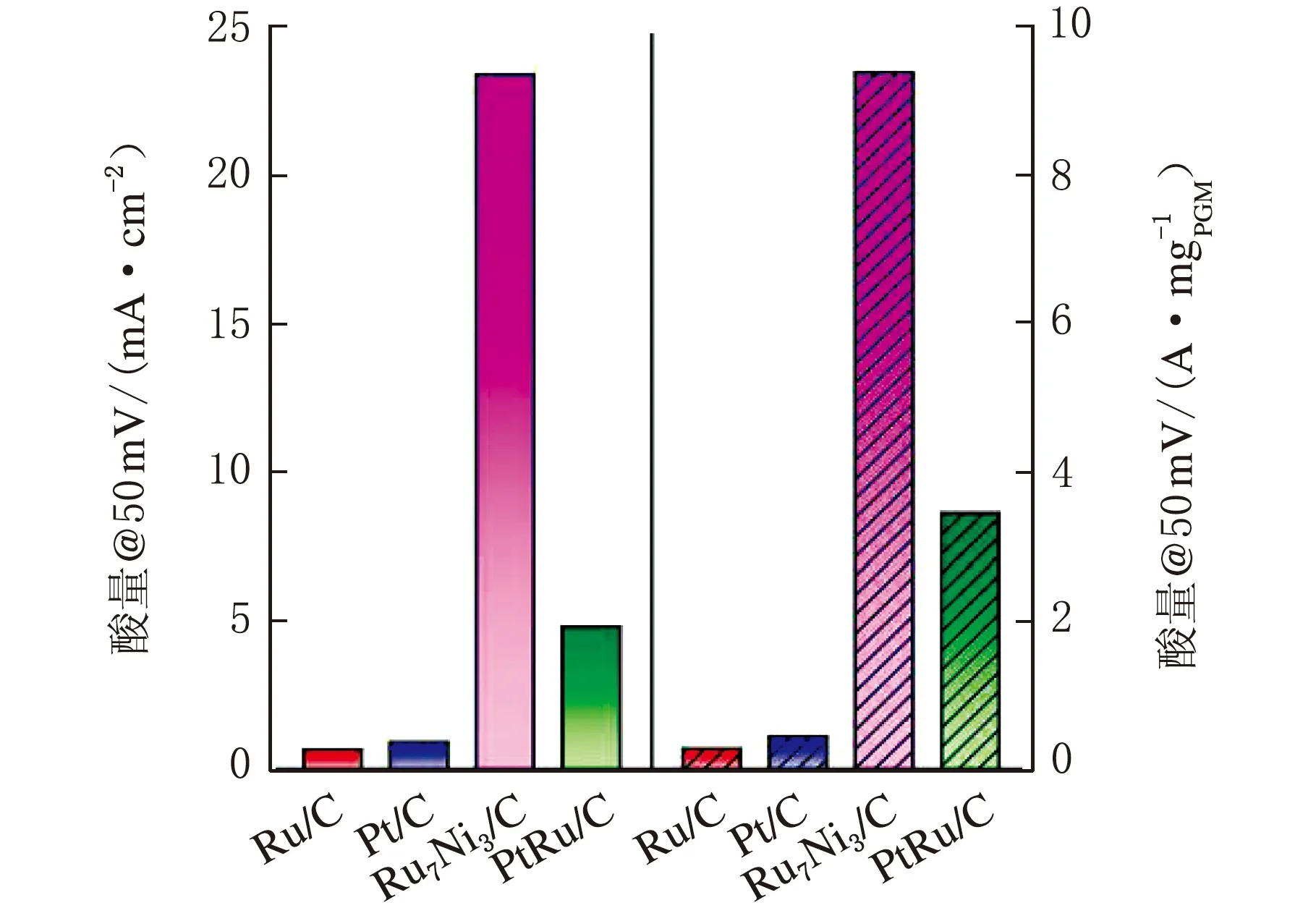
圖3 Ru/C、Pt/C、Ru7Ni3/C和PtRu/C電催化劑在50 mV vs.RHE處的質量比活性和面積比活性對比圖
基于上述的研究表明,Ru與其他金屬的合金化可以有效地調變Ru的電子結構,從而有效提高了Ru基電催化劑的堿性HOR活性。
2.2 核殼結構Ru基電催化劑
為了能夠實現減少電催化劑中貴金屬的負載量,以降低成本的同時還能保持高的電催化活性和穩定性,研究者們提出了核殼結構電催化劑。核殼結構納米顆粒是由至少2種相同或不同的物質組成的復合顆粒,其中一種物質成為核,另外一種物質形成包裹在外面的殼[35]。特殊的顆粒結構能夠很好地保留核心金屬原有的性能,同時還可以具有外層金屬的特性。這種復合材料能夠很好地克服單一金屬材料固有的缺陷,使得電催化性能和穩定性得到很大地提升,同時也能有效地解決貴金屬負載量高導致的成本問題。
Xing[36]課題組提出使用欠電位沉積(UPD)方法在Ru納米顆粒上逐層生長Pt層來調節表面性能,經過重復的UPD工藝獲得Ru NPs上的Pt多層結構,以此形成以Ru為核、Pt為殼層的核殼結構電催化劑。從Butler-Volmer圖上可以獲得Ru-rGO、Ru-MC(Pt原子單層沉積在Ru NPs) Pt-rGO、Ru-DC(雙層) Pt-rGO、Ru-TC(三層) Pt-rGO和Ru-QC(四層) Pt-rGO電催化劑的j0,s值分別為0.27、1.49、2.41、1.76和1.67 mA/cm2。顯然,所有合成的Ru@Pt核殼電催化劑都有著比Ru-rGO更好的電化學活性。運用循環伏安法(CV)研究M-H鍵能,結果表明所制備的一系列核殼結構電催化劑的UPD-Hdes的峰位置較Pt/C發生了負移,表明Ru能夠降低Pt-H的鍵能,也在某種程度上解釋了Ru@Pt核殼NPs優異的電催化性能。同時選用了CO溶出伏安法來探究親氧性,結果發現Ru-TC Pt-rGO和Ru-QC Pt-rGO上的CO溶出峰隨著Ru核上Pt厚度的增加而正向移動,交換電流密度幾乎隨表面親氧性的降低而線性降低。基于這2個結果提出HOR活性是通過雙功能機理實現的。其中Ru提供了吸附OH的活性位點,而附近的Pt原子為Had提供了活性位點而且Pt殼原子與Ru核原子之間的電子相互作用也可能增強了HOR活性。
2.3 Ru-金屬氧化物電催化劑
近幾年研究者們把目光逐漸轉移到了金屬-金屬氧化物電催化劑上,因為部分金屬氧化物在高電位、堿性條件下具有良好的穩定性。此外,金屬氧化物與金屬電催化劑之間強的相互作用可以在一定程度上改變金屬表面的電子結構從而改變金屬電催化劑的電催化活性,提高金屬電催化劑的穩定性。
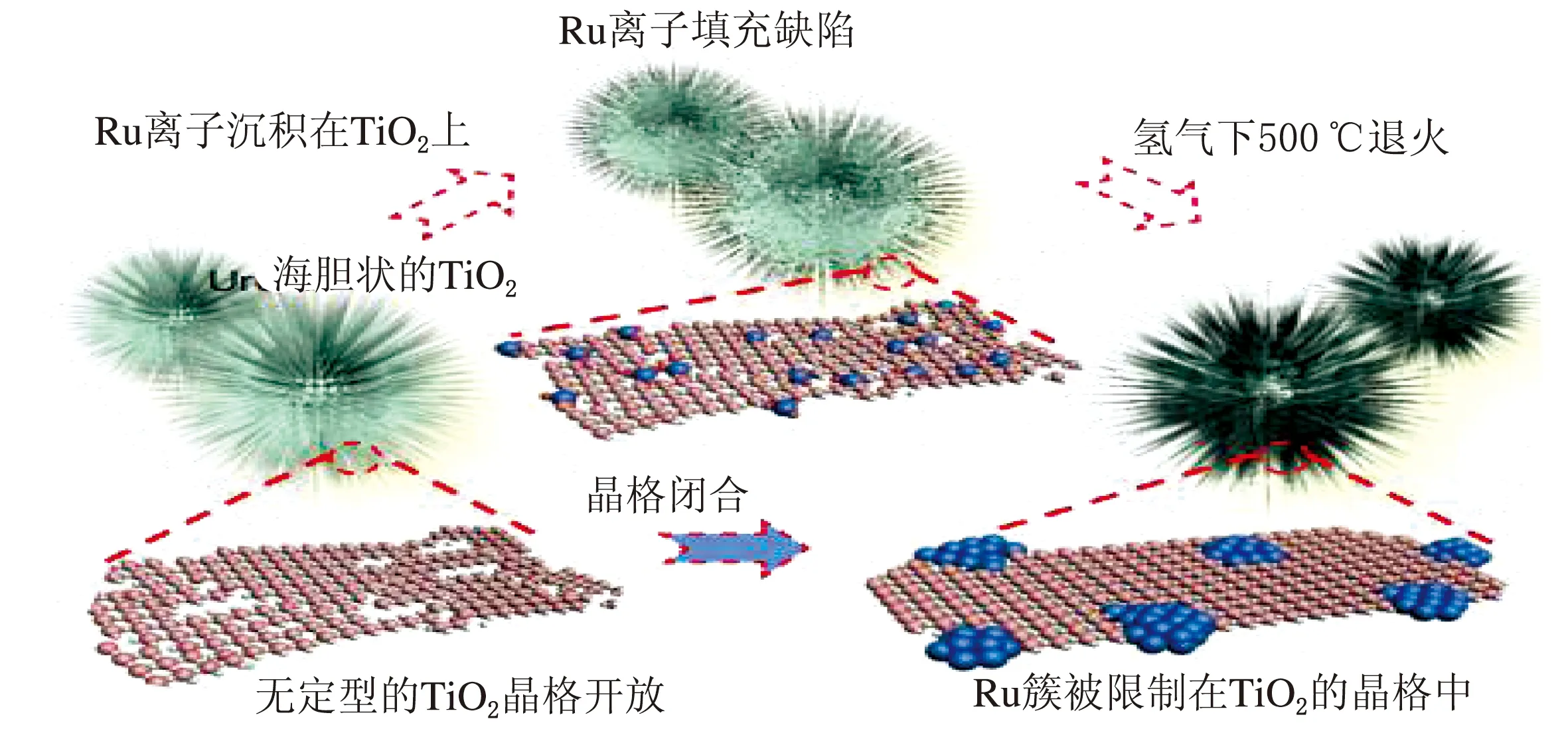
圖4 Ru@TiO2電催化劑的合成示意圖[37]
Jiang[38]等提出了相間氧化的概念(見圖5),Ru被TiO2相間氧化,2 nm的Ru簇可以在高達0.9 V(vs.RHE)的電位下有效地催化HOR,而自身不會被過度氧化。所制備的相間氧化電催化劑IO-Ru-TiO2/C的質量比活性為907 A/gRu@0.05 V vs.RHE,分別是Ru/C和PtRu/C電催化劑的17.5和1.5倍,其面積比活性為1.13 mA/cm2,分別是Ru/C(0.12 mA/cm2)和Ru-TiO2/C(0.15 mA/cm2)近9.4倍和7.5倍。
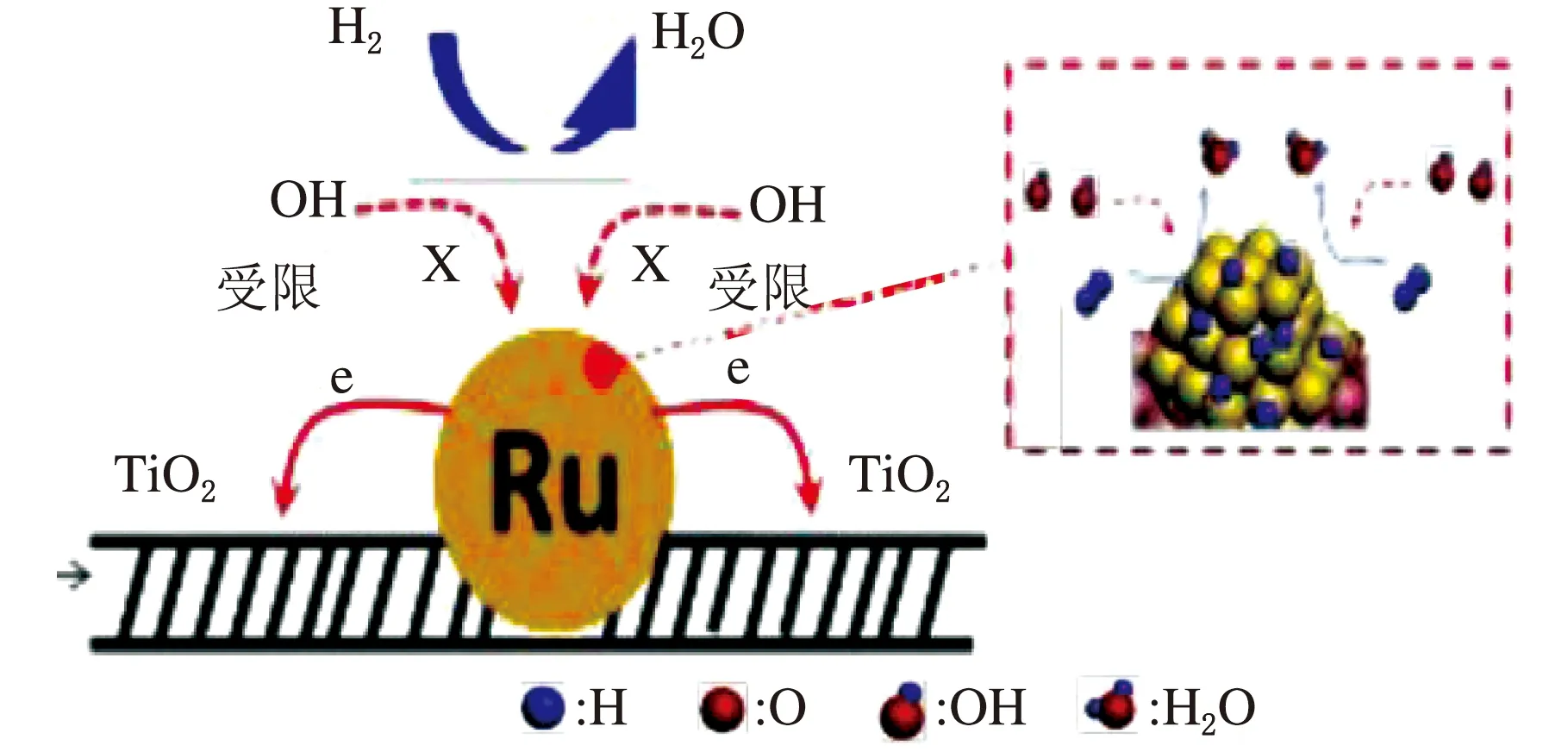
圖5 IO-Ru-TiO2/C相間氧化電催化劑的合成示意圖
活性的提高要歸因于形成了豐富的Ru-TiO2界面,界面上產生的電子轉移將Ru簇氧化為近似Ru4+,從而形成了相對穩定的半填充的4d軌道。這就使得Ru的進一步氧化變得困難從而保留了金屬表面。
2.4 載體優化的Ru基電催化劑
一般來說,載體本身是不具有電催化性能的,載體主要用于支持活性組分,使電催化劑具有特定的物理性質。合適的載體能夠使制得的電催化劑具有合適的形貌、尺寸等,也可以使電催化劑活性組分均勻分散在載體表面,以此獲得更高的電化學活性比表面積,提高單位質量電催化劑的催化效率[39]。載體的不同可以使電催化劑的性能有很大的差異,因此對于目前來說優化電催化劑的載體也是解決目前電催化領域問題的方法之一。
Zeng等[40]設計了一種具有中孔溝狀通道的介孔碳載體(Meso C)負載金屬Ru電催化劑。這種優化了載體結構的Ru/Meso C電催化劑有著十分優異的堿性HOR活性,其質量比活性為0.54 mA/μgRu是Ru/C(0.19 mA/μgRu)的2.8倍,其峰值功率密度為1.02 W/cm2,是Ru/C(0.76 W/cm2)的大約1.3倍,甚至可以在相同金屬載量下與商用Pt/C(20wt%)的單池性能相媲美。其課題組是為數不多的通過設計親水和疏水界面優化堿性HOR電催化劑的性能,所涉及的介孔碳載體有著中孔微觀結構,一方面能夠形成外部親水且內部疏水表面,從而確保了H2吸附和解離的理想界面環境,促進了氫的氧化。另一方面,多孔通道可以充當密閉空間,以防止內部預先錨定的Ru NPs過度暴露于空氣,從而保留比Ru/C更高比例的Ru金屬態。這種有效提高HOR活性降低貴金屬負載量的方式也為日后電催化劑設計提供了新思路。
Brkovic等[41]結合氧化鎢和碳化鎢的優點設計出了一種新型的電催化劑載體——非化學計量的碳化鎢氧化物(WxCyOz),主要是由非化學計量的氧化鎢(WOx)和碳化鎢(WC)組合而成,將其用作Pt-Ru納米顆粒的載體。在研究中發現質量分數為30%金屬載量的Pt-Ru/WxCyOz有著比商業Pt/C更好的電催化活性,在30 mV的HOR過電位下,質量分數為30%的Pt-Ru/WxCyOz表現出8.33 mA/cm2的動力學電流密度(jk)和0.028 A/mgPt的質量比活性,這些性能均優異于商業Pt/C(7.86 mA/cm2和0.197 A/mgPt)。
3 結束語
首先闡述了堿性HOR的作用機理,隨后著重總結了如今運用于堿性HOR的Ru基電催化劑,主要包括了Ru-M合金電催化劑、核殼結構Ru基電催化劑、Ru-金屬氧化物電催化劑以及載體優化的Ru基電催化劑。盡管近幾年來堿性HOR電催化劑的研究取得了很大的進展,但是就目前來說依然面臨著諸多挑戰。
HBE作為堿性HOR的重要指標之一,其很難在電化學條件下直接獲得,因為不是所有的金屬都具有不受干擾的氫吸附/脫附區,部分金屬的氫區涉及OH-的吸附。此外,OH-的作用也有待商榷,因此需要其他更實際有效的光譜方法在電催化條件下分析電催化劑上的HBE以及親氧性與活性之間的關系。
盡管使用Ru基電催化劑能夠降低陽極電催化劑的成本,但是Ru容易氧化,在電位較高的情況下,吸附的含氧物種會抑制氫的吸附,降低堿性HOR活性。Xue[42]等人曾在其文章中指出在實際應用時,HEMFCs可能會出現偏離正常陽極工作條件的情況,比如電池啟動和關閉時,陽極會暴露在較高的電位下同時可能會出現反向電流,這就對陽極電催化劑在高電位條件下仍需具有穩定高效催化HOR有了更高的要求。
為了更好地研究和設計堿性HOR電催化劑,可以使用原位技術實時觀測堿性HOR的催化過程,探索堿性HOR反應中間體,這有助于明確電催化劑的真實催化活性位點,為開發高性能的電催化劑提供理論指導[14]。此外,可以借助密度泛函理論認識電催化劑活性位點的最優結構,進一步優化電催化劑的制備參數,解決高電位條件下Ru易氧化問題,構筑具有高交叉頻率、高穩定性活性位點的Ru基堿性HOR電催化劑。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
