LC-MS/MS測定風味發酵乳中的矢車菊素-3-葡萄糖苷和矢車菊素-3-蕓香糖苷
2021-02-03 11:47:04*李麗
當代化工研究 2021年24期
*李 麗
(江蘇聯合職業技術學院鎮江分院 鎮江高等職業技術學校 江蘇 212000)
引言
矢車菊素-3-葡萄糖苷(鹽酸鹽分子式C21H21O11Cl,CAS:7084-24-4,圖1)和矢車菊素-3-蕓香糖苷(鹽酸鹽分子式C27H31O15Cl,CAS:18719-76-1,圖2)屬于黃酮類物質,在黑加侖、桑椹等植物產品中廣泛存在[1]。

圖1 矢車菊素-3-葡萄糖苷鹽酸鹽
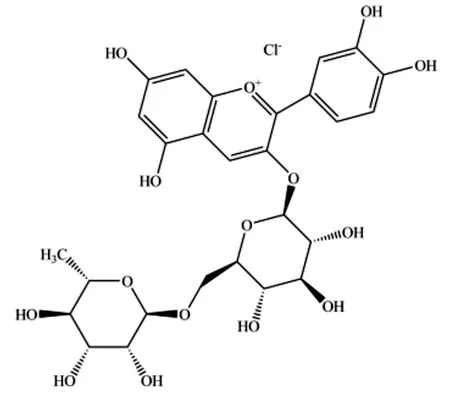
圖2 矢車菊素-3-蕓香糖苷鹽酸鹽
對于桑椹紅作為食品著色劑的使用,GB1886.345-2021《食品安全國家標準 食品添加劑 桑椹紅》[2]給出了兩種分子結構式:矢車菊素-3-葡萄糖苷,[C21H21O11]+X-;矢車菊素-3-蕓香糖苷,[C27H31O15]+X-,其中X-為平衡離子。雖然C3G、C3R具有良好的健康功效[3],但是在GB2760-2014《食品安全國家標準食品添加劑使用標準》[4]的A.2表中,未將其列入可在各類食品中按生產需要適量使用的食品添加劑范疇。A.1表只規定了果糕類、糖果等六類食品中允許添加桑椹紅色素。日常食用的風味發酵乳并沒有在允許使用的品種之列。
目前,C3G和C3R的檢測有高效液相色譜法(HPLC)[5-7]和LC-MS/MS法。HPLC法屬于常規的定量方法,關注的是草莓、黑加侖等產品中這兩種化合物的含量,C3G和C3R的濃度相對較高易于測定。但HPLC測定C3G和C3R存在以下的缺陷:(1)參考國際通行的做法,對于禁用物質的檢測,液相色譜的測定結果不易作為最終的判定依據,需要配合質譜、紅外、拉曼、核磁等技術手段對分子結構進行定性或者更換不同的色譜柱予以確認。(2)樣品前處理的方法或者色譜分離條件不適用于質譜的分析。在上述HPLC方法中,樣品待測液中含有高濃度的無機酸(如磷酸)或者流動相中使用了鹽酸,無機酸的存在會嚴重污染或者損害質譜檢測器。Janine M Cooney[8]等采用直接過SPE柱的方式,用LCMS/MS測定了尿液中C3G和C3R的含量,樣品基質相對簡單,易于處理;Chunxia Yang等用LC-MS/MS測定了血清中C3G的含量,在樣品前處理過程中待測組分的損失較大,需要用槲皮素-3-葡萄糖苷作為內標進行校正,該方法能否適用風味發酵乳的測定尚待評估。
針對風味發酵乳中含有蛋白、糖、甜味劑、增稠劑、乳化劑等物質的特點,本研究擬采用乙腈、甲醇、乙酸鉛、三氯乙酸等不同的溶液提取待測組分,并對其提取效率和凈化方法進行評估和確認,為風味發酵乳中C3G和C3R的定性定量測定提供了技術方面的參考。
1.材料與方法
(1)儀器及試劑
Thermo TSQ Quantan Access液相色譜-串接質譜儀(配電噴霧離子源(ESI)和自動進樣器,美國Finnigen公司);XPE205電子天平(瑞士梅特勒公司);3K15高速離心機(德國Sigma公司)。
甲醇、乙醇、正己烷(色譜純,德國Merck公司);甲酸、氨水、乙酸鉛、三氯乙酸(分析純,上海國藥集團);實驗用水采用高純水(美國Millipore公司);水相膜(0.22μm,上海安譜公司);Poly-Sery MCX固相萃取柱(500mg 6mL,上海安譜公司);C3G標準品(CAS 7084-24-4)、C3R標準品(CAS 18719-76-1)(純度均大于≥98.0%,成都草源康生物科技有限公司)。
(2)實驗方法
①標準溶液的配置
稱取C3G和C3R標準品各5.0mg(±0.1mg)于15mL棕色玻璃瓶中,加入10.0mL水(含0.4%甲酸)溶解振蕩,配制成濃度為500mg/L的標儲。冷凍(-18℃)密閉暗室保存。根據后續需要加0.2%甲酸水稀釋。
②制樣
取同一批次的風味發酵乳4盒,用組織搗碎機混勻,待用。
③樣品前處理過程
A.提取待測組分。稱樣2.00g(±1.0mg)于15mL具塞離心管中,加10mL 10%三氯乙酸溶液渦旋10s,超聲處理8min。冷凍離心25min(8000r/min,R=8cm,-4℃),取上清液加3ml正己烷渦旋1min,離心3min,棄去正己烷層,水相待凈化。
B.凈化。Poly-Sery MCX柱先后用5mL甲醇、5mL 0.2%甲酸水清洗,保持柱床的濕潤。加入5mL提取液過柱。用5mL甲醇和5mL 0.2%甲酸水溶液清洗柱床。用洗耳球將柱床吹干,加入8mL 15%氨水-甲醇洗脫,收集全部濾液氮吹至200μL左右,補加0.4%甲酸水至2.0mL,采用渦旋的方法再次溶解,過水相膜待測定。
(3)液相色譜-串接質譜檢測條件
①液相色譜條件
Agilent Eclipse Plus C18柱(150mm×2.1mm,3.5μm);流動相C:乙醇(含0.05%甲酸),流動相D:水(含0.2%甲酸),流動相A:甲醇,梯度洗脫見表1。流速0.25mL/min;進樣量20μL;室溫。

表1 色譜梯度洗脫條件
②質譜條件
電噴霧離子源(ESI),正離子模式掃描;質譜掃描方式:多反應監控(MRM);噴霧電壓3800V;傳輸毛細管溫度400℃;輔助氣流量2.5mL/min;殼氣流量10mL/min。
2.結果與分析
(1)質譜條件的優化

續表
將標準溶液配成10.0mg/L的濃度,在正離子模式下進行母離子、子離子的優化。C3G、C3R的監測量離子對、能量、保留時間見表2。標液的總離子流圖和質譜見(圖3、圖4)。

圖3 C3G的總離子流圖和質譜圖(濃度3.125μg/L)

表2 待測組分的質譜參數、保留時間

圖4 C3R的總離子流圖和質譜圖(濃度3.125μg/L)
(2)樣品提取和凈化條件的選擇
①在空白樣品中加入相同濃度的混合標準溶液情況下(50μg/kg),考察了0.1%甲酸乙腈、0.1%甲酸乙腈+甲醇(乙腈:甲醇=2:1,v/v)、20%乙酸鉛、三氯乙酸作為常用的蛋白沉淀劑的沉淀效果以及提取效率。用0.1%乙腈提取待測組分,沉淀效果好、上清液澄清,但是C3G、C3R的回收率分別只有63.1%和60.2%;用0.1%乙腈+甲醇提取待測組分,C3G、C3R的回收率分別為44.1%和25.2%;20%乙酸鉛的提取效率相對較差,C3G、C3R的回收率分別只有3.58%和4.52%,可能跟高濃度的乙酸鉛中的離子占據了MCX的吸附點位有關。低濃度的三氯乙酸沉淀效果不佳,會產生比較大基質干擾,回收率偏高。經綜合測試,采用10%的三氯乙酸處理樣品可以取得良好的效果。同等濃度的C3G加標測定值的峰面積大小順序依次為:10%的三氯乙酸>乙腈>0.1%甲酸乙腈+甲醇>20%乙酸鉛(圖5);C3R的測定結果與C3G一致(圖6)。

圖5 四種溶劑處理加標樣品后,C3G的總離子流圖

圖6 四種溶劑處理加標樣品后,C3R的總離子流圖
②在色譜的分離階段,選擇了甲醇、0.05%甲酸甲醇、乙醇、0.05%甲酸乙醇作為洗脫溶液。試驗中發現用甲醇洗脫C3G和C3R,會產生少量的柱殘留。采用0.05%甲酸乙醇洗脫待測組分,不僅可以消除柱殘留現象,也有利于待測組分響應值的提高。
(3)方法的標準曲線、線性范圍、檢出限和定量限
吸取100μL標準混合溶液(1000μg/L)于900μL 0.2%甲酸水溶液中,渦旋混勻。再將混合溶液用0.2%甲酸水倍比稀釋至6個濃度:3.12μg/L、6.25μg/L、12.5μg/L、25.0μg/L、50.0μg/L、100μg/L。以定量離子的峰面積為橫坐標,質量濃度為縱坐標,繪制工作曲線。所得工作曲線線性良好,相關系數R2大于0.995(圖7、圖8)。

圖7 C3G標準曲線圖(濃度3.12μg/L-100μg/L)
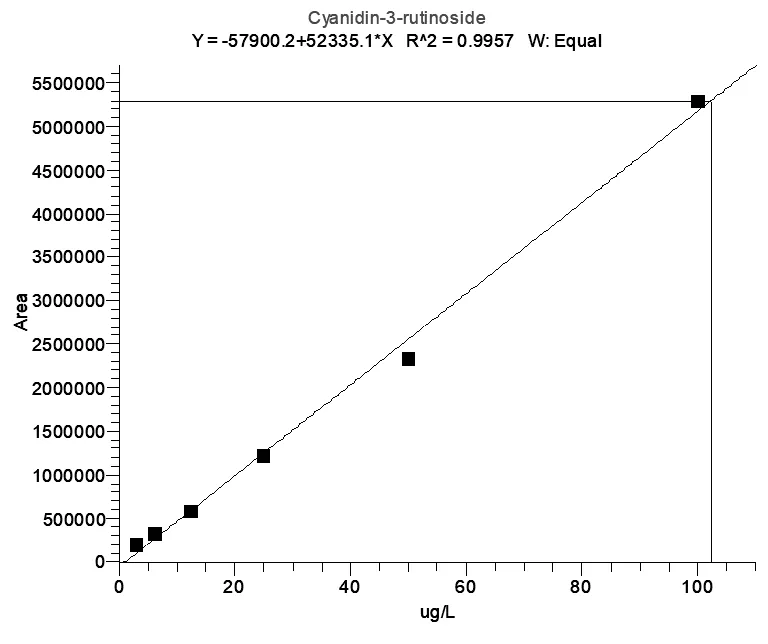
圖8 C3R標準曲線圖(濃度3.12μg/L-100μg/L)
參照Eurachem的方法[8],計算出這兩種化合物檢測方法的檢出限和定量限均為2.50μg/kg、5.00μg/kg。
(4)回收率及精密度
空白樣品進行加標回收檢測,添加水平:5.00μg/kg(LOQ)、10.0μg/kg(2×LOQ)、50.0μg/kg(10×LOQ),每個平行測定6次。C3G的加標回收率為103%~115%,相對標準偏差(RSDs)為4.17%~5.88%;C3R的加標回收率為105%~119%,相對標準偏差5.32%~6.07%。空白樣品、加標樣品總離子流見(圖9、圖10)。

圖9 空白樣品的總離子流圖
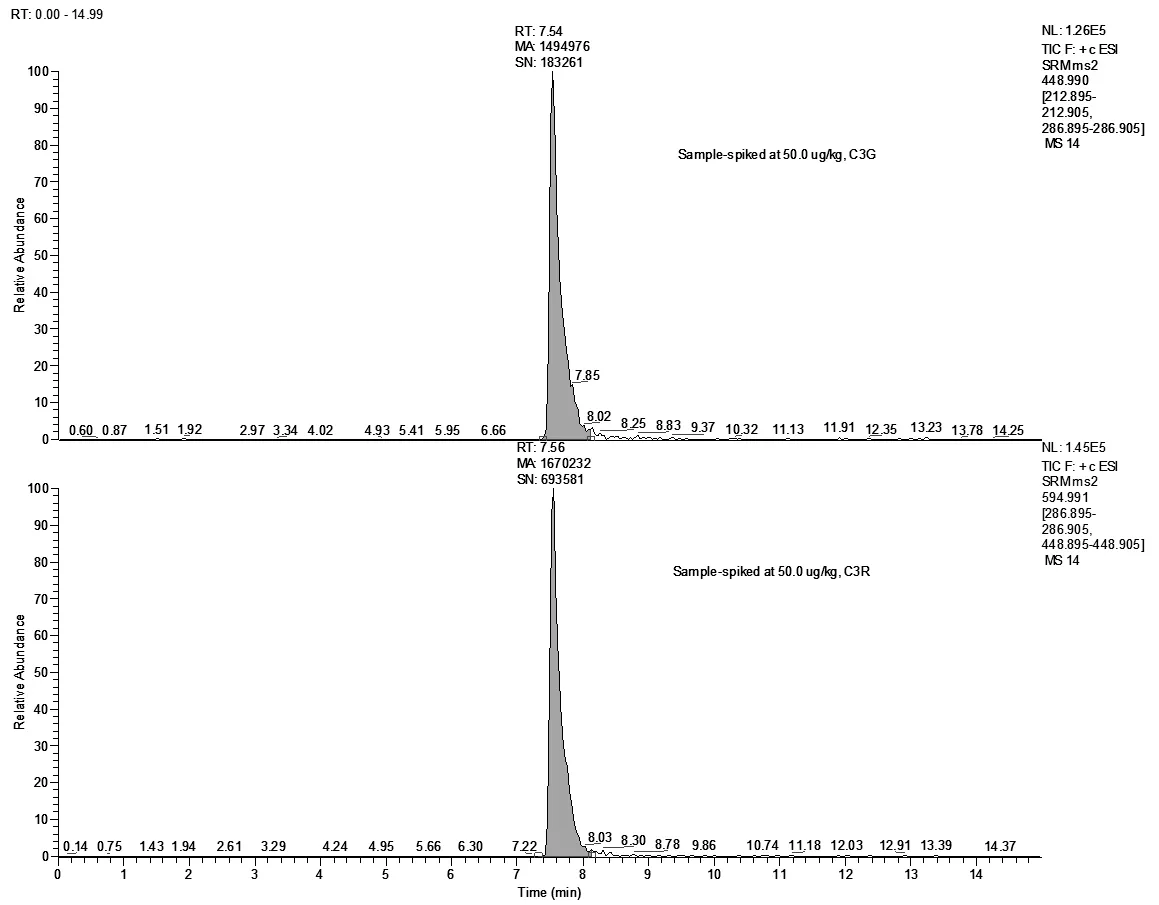
圖10 發酵乳加標樣品的總離子流圖(加標濃度50.0μg/kg)
(5)樣品的篩查
原味、紅棗味等8份從隨機采購的發酵中都沒有檢出。
3.結論
風味發酵乳經三氯乙酸沉淀蛋白并提取純化,把色譜洗脫條件及儀器參數進行調整優化后,建立了液-質連用檢測C3G、C3R的檢測方案,應用于市面上常見的發酵乳的檢測。所測的8份隨機購買的發酵乳,都沒有檢測出上述兩種物質。該方法的建立為將來更加合理、科學地制定C3G、C3R的使用標準,防止該類色素的不規范使用,提供了技術支撐。
