架起化學-酶催化之間的橋梁:構建策略及催化應用
2021-01-29 08:01:22欒鵬仟周丹丹王曉天陳冉高士耆趙浩黃琛劉運亭高靜姜艷軍
化工學報 2020年12期
欒鵬仟,周丹丹,王曉天,陳冉,高士耆,趙浩,黃琛,劉運亭,高靜,姜艷軍,2
(1 河北工業大學化工學院,天津300130; 2 河北工業大學化工節能過程集成與資源利用國家地方聯合工程實驗室,天津300130)
引 言
化學催化在催化領域一直占據著主導地位,尤其在精細化學品[1-3]、先進材料[4-6]、電池[7-9]等領域得到廣泛應用,為人類社會進步做出極大的貢獻。然而,化學催化仍然存在一些不可避免的缺點,如環境污染、選擇性差、反應條件苛刻等。近年來,生物催化以其反應條件溫和、環境友好以及(化學、區域和立體)選擇性強等優點成為研究熱點,在一定程度上能夠彌補化學催化的不足[10-11]。盡管如此,生物催化同樣也存在著許多局限,其中最為突出的是生物催化劑的穩定性、催化反應的多樣性、重復使用性等都遠不如化學催化劑,并且制備條件復雜,周期長。將化學催化與生物催化相結合,實現兩者的優勢互補,并用于設計全新、精巧的反應來合成高附加值產品,逐漸成為新的研究領域[12-13]。自1997 年B?ckvall 等[14]首次將水解酶和金屬催化劑結合在一起,化學-酶級聯催化至今已經取得了極大的進步。
傳統的化學-酶催化轉化通常是逐步進行的,即完成第一步反應后需要通過復雜的分離純化技術將中間產物從體系中分離,然后才能進行接下來的反應。這種方式的化學-酶催化不僅費時費力,而且增加了整個工藝的成本。為解決這些弊端,研究者開發了一鍋法化學-酶級聯催化反應,所有反應均在同一個容器中進行,省略煩瑣的分離純化步驟,從而簡化工藝降低成本[15-17]。
盡管工業上使用化學-酶催化轉化的實例正在不斷增長,但其內在的復雜性嚴重阻礙了這一概念的廣泛應用,其中最大的障礙是化學催化與生物催化的不相容性。這種不相容性包括不同催化劑自身的不相容和催化條件差異導致的不相容。催化劑自身的不相容性主要表現在一鍋法轉化中化學催化劑與酶的直接接觸導致催化劑相互失活。例如,Latham 等[18]在結合鹵化酶和金屬鈀(Pd)的研究中發現,酶的氧化還原性和親和力會嚴重影響Pd的活性。而反應條件的差異性表現得更為明顯,酶催化一般需要在條件較為溫和的水相介質中發生,而大多數化學催化劑不僅要在高溫強堿下發揮作用,而且還需有機溶劑作為反應介質。在這種苛刻的環境下,大多數的酶都很難維持原有的催化活性,例如酚酸脫羧酶(PAD)在疏水溶劑中是有活性的,但是長時間暴露在甲苯溶劑中會導致其完全失活[19],而甲苯正是大多數化學催化劑較為理想的反應介質。化學催化劑和生物催化劑任何一方的失活都將直接導致化學-酶催化轉化的失敗,因此需要發展有效的策略來應對不同原因導致的不相容。
基于以上背景,本文總結了克服化學催化與酶催化不相容問題的方法和策略,包括時間分隔和空間分隔策略。另外,還介紹了化學-酶級聯催化在手性化合物動態動力學拆分和藥物分子(或中間體)合成等方面的應用。最后,對未來化學-酶級聯催化的發展方向進行展望。
1 化學-酶級聯催化構建策略
催化條件(如溫度和pH)引起的不相容,決定了兩種催化反應不能同時發生。因此一般采取時間分隔的方式,第一步反應結束后,調節溫度和pH 到下一步反應所需的范圍后,再加入催化劑與試劑進行后續反應。對于溶劑帶來的問題可采用兩相體系和非常規溶劑的方法來解決。催化劑不相容的問題通常采用空間分隔的策略來解決,具體方法包括將不同催化劑各自固定化、制備分隔式集成催化劑或借助高分子膜來分隔催化劑。通過這些手段均可有效實現不同催化劑的活性中心在空間上的分離,從而避免接觸失活。
1.1 催化條件不相容
1.1.1 時間分隔(temporal separation) 一鍋化學-酶級聯反應分為三類,分別為連續、并行和協同反應。在反應過程中需要調節體系溫度、pH、底物濃度等條件的反應為典型的連續反應。與其他兩類反應相比,連續反應過程需要進行額外的操作,譬如添加反應組分或調節反應條件等,使得過程更加煩瑣,但是該方法可以將更多催化條件相差很大的催化劑級聯在一起,創造出更多全新的一鍋化學-酶催化反應,因此應用最為廣泛。
González-Sabín 等[20]通過連續反應的方式實現了釕(Ru)基催化劑和轉氨酶(ω-TA)的結合。如圖1(a)所示,Ru催化的烯丙醇異構化在50℃條件下進行,而ω-TA 卻不能耐受50℃的高溫條件。此外,在研究中發現底物烯丙醇對轉氨酶的活性存在一定程度的影響。因此,在化學催化完成之后,先降低溫度到轉氨酶耐受的范圍,將體系稀釋使底物濃度降低從而減小對酶活的影響,最后再將轉氨酶引入體系,進行后續的生物催化。由此可看出采取連續反應的方式進行一鍋化學-酶催化轉化可以有效地消除催化條件差異帶來的不相容問題。
金屬催化的C—C 交叉偶聯反應已被公認為有機化學中形成C—C 鍵最有力和最通用的合成工具之一,如Suzuki、Heck 和Negishi 反應。這類反應最吸引人的一個優點在于其可在水相中進行,有利于與生物催化的結合。Groger 等[21]首次將Pd 催化的Suzuki交叉偶聯反應與醇脫氫酶(ADH)催化的酮還原反應相結合,并成功地在水相中完成了二者的級聯反應,如圖1(b)所示。在70℃的堿性水溶液中,Pd(PPh3)Cl2催化苯硼酸與對溴苯乙酮發生C—C 交叉偶聯反應。反應結束后無須分離中間產物,待體系溫度降為室溫、調節pH 到中性后加入ADH 進行后續的還原反應,最終生成一系列手性二芳醇。Garg等[22]用類似的方法在水中級聯制備抗阻胺藥物奧非那汀的對映體。Turner 等[23]開發了一種一鍋化學-酶級聯反應,采用了時間分隔的連續反應方式,最終得到一系列光學純的N-保護的非天然L-和D-二芳基丙氨酸衍生物。
除了前面介紹的交叉偶聯反應外,González-Sabín 等[24]采用連續反應的方式成功地在水相中將KRED 催化的不對稱還原反應與金屬催化的炔烴環異構化反應相結合,以較高的產率合成了1,4-二醇、內酯、γ-羥基羰基化合物(羧酸、酯和酰胺)等多種對映體純化合物,如圖2(a)所示。同年該課題組又建立了外消旋醇不對稱轉化為胺的化學-酶級聯催化體系[25],如圖2(b)所示。將有機催化劑2-氮雜金剛烷-N-氧自由基(AZADO)、氧化劑次氯酸鈉(NaOCl)和轉氨酶結合,在水介質中實現了一鍋氧化-胺化連續過程。該方法具有寬廣的底物譜,包括傳統的仲醇和空間位阻較大的β-取代環烷醇。其中高度立體選擇性的不對稱生物胺化使整個反應過程具有非常高的產率、光學純度和非對映選擇性(產率>90%,ee>99%,dr>49∶1)。
化學-酶級聯反應中催化劑的不相容性及其交叉反應性問題頻繁出現,使得大多數例子都是連續的一鍋反應,而不是真正的并行反應。但是連續反應也存在一些局限性,比如反應過程中涉及可逆反應或極不穩定的中間體時,該方法則不適用。因此,需要發展其他的策略,以解決這些問題或實現更為高效的并行反應。
1.1.2 兩相體系 除了溫度和pH 引起的催化條件不相容外,溶劑引起的不相容性同樣不可忽視。絕大多數化學催化劑偏好有機介質,在水相中很難正常發揮催化活性,但幾乎所有的酶對有機溶劑是不耐受的。而且,有機試劑在水相中的溶解度普遍較差,這大大增加了使用水作為反應介質的難度。因此,研究者利用兩相體系解決溶劑帶來的不相容問題以及底物溶解性問題[26-27]。該體系由兩種互不相溶的液體組成,化學催化劑溶解在有機相中,而酶溶解在水相中,從而有效地緩解溶劑帶來的不相容問題。從另一個角度上考慮,兩相體系在一定程度上也能限制不同催化劑之間的接觸,避免催化劑發生接觸失活。
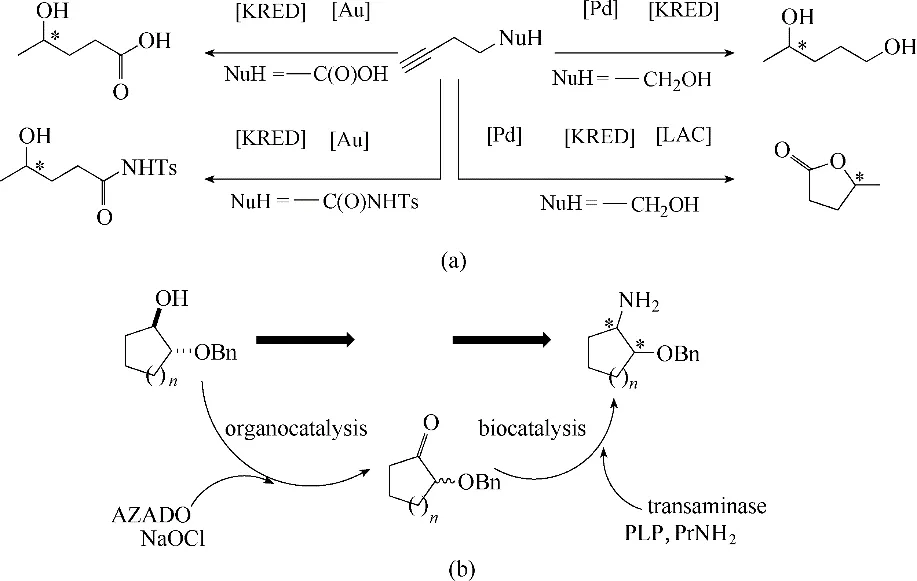
圖2 水溶液中化學-酶催化不對稱級聯合成1,4-二醇、內酯、γ-羥基羰基化合物[24(]a)及2-(芐氧基)環烷胺[25(]b)Fig.2 Chemoenzymatic for the asymmetric synthesis of 1,4-diols,lactones,γ-hydroxy-carbonyl compounds[24](a)and(benzyloxy)cycloalkanamines[25](b)in aqueous medium
Castagnolo 等[28]開發了一種由水和異辛烷組成的兩相體系用于一鍋化學-酶催化合成吡咯,如圖3(a)所示。首先底物和Ru 基催化劑溶于異辛烷中,酶溶于緩沖溶液中。隨著化學催化的不斷進行,生成的中間產物3-吡咯啉不斷地向緩沖溶液中擴散,隨即在單胺氧化酶(MAO-N 或6-HDNO)的作用下發生芳構化反應,從而在水相中生成吡咯。而且,生成的吡咯又不斷地從水相擴散到有機相中,促進了反應平衡向產物方向的移動,這是該反應在溫和條件下高產率地合成吡咯的原因。實驗結果表明該兩相體系解決了溶劑引發的不相容問題,而且還避免了化學催化劑與酶發生絡合作用。最近,該課題組又應用該兩相體系,以脂肪族二烯丙基醚為原料,結合Ru基催化劑催化的烯烴復分解反應和漆酶(laccase)催化的芳構化反應,通過一鍋法化學-酶級聯催化合成了一系列呋喃類化合物[29],如圖3(b)所示。由異辛烷與緩沖溶液組成的穩定的兩相體系還被應用于其他一鍋化學-酶級聯反應中[30-31]。
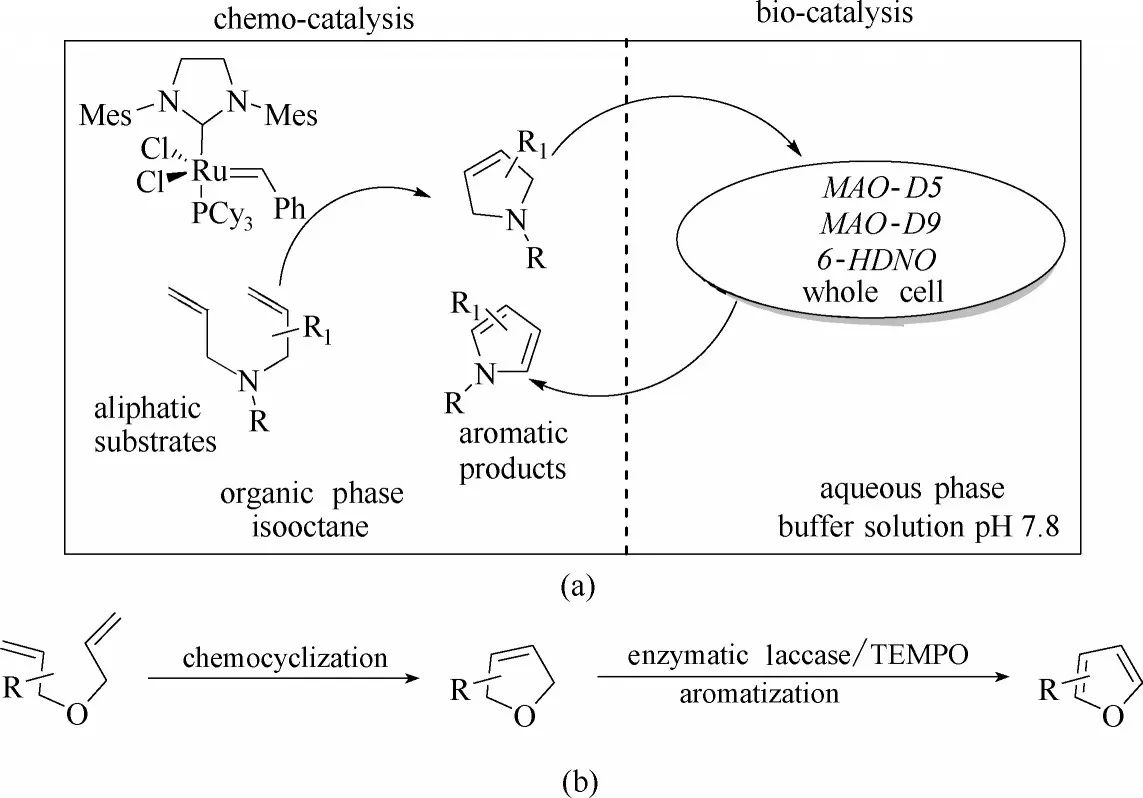
圖3 兩相體系一鍋化學-酶催化合成吡咯[28](a)和呋喃[29](b)類化合物Fig.3 One-pot chemoenzymatic synthesis pyrroles[28](a)and furans[29](b)compounds though biphasic system
蔗糖磷酸化酶(SP)能夠催化蔗糖可逆地磷酸化為α-D-葡萄糖-1-磷酸和D-果糖,其廣泛的受體特異性被開發用于葡萄糖向多種受體分子轉化,如多元醇、酚類、羥基呋喃酮和二苯乙烯類化合物。然而由于SP 對這些底物的低親和力(Km>1 mol/L),通常情況下需要向體系中加入二甲亞砜或甲醇等助溶劑來提高底物的溶解度。助溶劑的濃度對反應影響很大,濃度過低時不足以溶解足夠的底物,而濃度過高時會直接抑制酶的活性甚至使其失活。Winter 等[32]利用由緩沖溶液和乙酸乙酯組成的兩相體系通過化學-酶法合成α-D-葡萄糖苷。水相中含有酶和水溶性底物,而疏水性底物則溶解在有機相中。通過攪拌或搖動使這些底物由有機相轉移到水相,并在水相中發生酶催化轉化。通過對比產物2-O-α-D-葡萄糖苷鄰苯三酚在助溶劑體系和兩相體系中的轉化率,發現后者的轉化率提高了5 倍之多,有效地抑制了副產物的產生。
手性亞砜是一類具有生物活性的重要化合物,其中一些亞砜是市售藥物的活性成分,如奧美拉唑[33]。此外,它們還可作為不對稱合成中的手性配體、催化劑和構建模塊[34]。Mí?ek 等[35]在由緩沖溶液和正癸烷組成的兩相體系中首次開發了對手性亞砜進行去消旋化反應。在該反應體系中,酶可以耐受不同的取代基,底物的適用范圍較廣,為合成對映體純亞砜提供了一條可行的途徑。
PAD 是典型的在有機溶劑中失活的酶,而且催化的底物在水中溶解度極低。Kara等[36]將PAD的脫羧反應與Pd催化的對羥基苯乙烯氫化反應耦合,在由緩沖溶液和正己烷組成的兩相體系中合成4-乙基愈創木酚。然而這種傳統的兩相催化體系存在一個很明顯的缺陷,反應僅發生在表面積有限的液面之間,導致反應速率緩慢。解決這類問題最直接的方法是增大液面之間的接觸面積。Kourist等[37]將PAD 封裝于聚乙烯醇(PVA)/聚乙二醇(PEG)冷凍凝膠中與化學催化劑催化的烯烴復分解反應級聯制備具有強抗氧化活性的4,4′-二羥基二苯乙烯,如圖4(a)所示。封裝后的PAD 不僅可在叔丁基甲醚(MTBE)中正常進行催化,而且凝膠還增大了液面的接觸面積。底物從有機相進入水相,催化得到的中間產物重新進入有機相。分離PAD 凝膠后,添加化學催化劑進行后續的烯烴復分解反應。
如上所述,將酶封裝于“油包水”的結構中可以實現有機相中的酶催化反應。與之類似,將化學催化劑封裝于“水包油”的結構中可以實現水相中的化學催化反應。TPGS-750-M 表面活性劑在水中可形成內部疏水外部親水直徑約50 nm 的球形膠束,內部的疏水核由維生素E 組成,可裝載過渡金屬催化劑和親脂底物,如圖4(b)所示。反應發生在這些特制的納米反應器內部的疏水核心,并且生成的產品在膠束之間可通過水進行動態交換,因此存在與酶構建級聯反應的潛力。Lipshutz 等[38]利用TPGS-750-M 形成的膠束實現了金屬催化反應(Pd 催化Heck偶聯反應、Au/Ag催化炔烴水合反應和Rh催化1,4-加成反應)與生物催化反應(ADH 催化還原反應)的結合,合成了一系列光學純的手性仲醇。其中,金屬催化的有機反應發生在膠束內部,而酶催化發生在與膠束相容的水介質中。這類設計可有效地將金屬催化劑限制在有機相中,從而避免外部水相對金屬的干擾,同時也阻斷了酶與有機相的接觸,使酶能夠一直在水相中維持較高的活性。水中存在的納米膠束不僅可為化學和生物催化提供反應介質,而且還可作為底物、產物和催化劑的貯存器,起到降低非競爭性酶抑制的效果。
1.1.3 非常規溶劑(non-conventional media) 兩相體系在一定程度上可以緩解溶劑帶來的不相容問題,然而低產率和內在的復雜性嚴重限制了兩相體系在間歇反應中的應用。近年來,非常規介質的發展給上述問題帶來了新的解決方案[39]。非常規介質包括:無溶劑過程、超臨界流體(SCFs)、生物質衍生溶劑、氟化溶劑、離子液體(ILs)和低共融體(DESs)。本節將重點介紹非常規溶劑用于解決一鍋化學-酶催化過程中溶劑引起不相容問題的研究進展。
ILs 是由有機陽離子和有機或無機陰離子組成的低熔點鹽,研究表明ILs 能夠通過產生的微環境穩定不同種類的酶,并起到液體載體的作用。此外,使用ILs 作為反應介質,在一定程度上能夠增強生物催化劑的對映選擇性和催化劑活性,并使其具有更高的操作穩定性。Schmitzer 等[40]在由含ILs 和水相組成的兩相體系中,實現了Pd 催化的Suzuki偶聯與ADH 催化的生物還原反應的級聯。Suzuki 偶聯發生在ILs 相中,ILs 可增強金屬催化劑的催化活性和穩定性。生成的中間產物轉移到水/IL 界面上被ADH 還原,最終以高收率和高對映選擇性合成了一系列手性二芳醇。
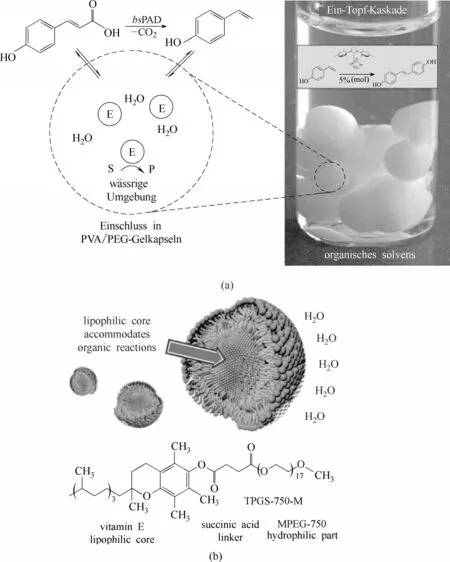
圖4 (a)PAD封裝于PVA/PEG凝膠中用于一鍋化學-酶催化反應[37];(b)TPGS-750-M在水相中形成的膠束[38]Fig.4 Encapsulate PAD in PVA/PEG for one-pot chemoenzymatic cascade reaction[37](a);The designer surfactant TPGS-750-M forms micelles in water[38](b)
DESs 是由廉價的生物可降解氫供體和氫受體組成的混合物,作為一種新型綠色溶劑在生物催化、金屬催化、有機合成和材料化學等領域得到了廣泛的應用。González-Sabín 等[41]首次將DESs 應用于酮的不對稱胺化中。ω-TA 在75%(質量)DESs和緩沖液組成的反應介質中具有很好的穩定性。將Pd 催化的Suzuki 交叉偶聯反應與異源外生菌ATA(EX-ωTA)催化的胺化反應級聯,實現了在該混合介質中合成雙芳胺。DESs 的增溶特性使金屬催化過程中底物的濃度和酶催化過程中中間產物的濃度分別升高至200 mmol/L 和25 mmol/L。與之前的研究相比,DESs的引入提高了產品12%的收率。并且ω-TA 在該混合溶劑中所展現出的超高酶活,無疑突出了生物可再生溶劑的實際應用價值。Wang等[42]通過在兩相體系中加入DESs,得到了穩定的微乳液。由于DESs降低了溶液的表面張力,加之脂肪酶的界面活化表現出的高活性導致產物的合成速率提高了六倍之多。
Capriati 等[43]將Ru 催化的烯丙醇異構化與KRED 的不對稱生物還原進行耦合,制備出一系列手性醇化合物。KRED 在由DESs 和緩沖溶液組成的混合溶液中表現出很好的穩定性與催化活性,而且研究發現混合介質中DESs 的含量越高,KRED 的對映選擇性越強,從而所得仲醇的光學純度越高。同年該課題組又以DESs 和緩沖溶液為反應介質獲得一組產率高、對映體純(ee>99%)的手性雙芳醇[44]。
1.2 催化劑自身不相容
除了不同催化條件引起的不相容,催化劑自身不相容也是建立化學-酶催化體系時常見的障礙。解決此類問題最常用的方法是將催化劑空間分隔(space separation),阻斷活性中心在空間上的接觸。空間分隔的方法分為:獨立固定化催化劑、分隔式集成催化劑和膜分隔。本節將對近年來研究者通過制備各種先進材料應用于化學-酶催化反應中解決催化劑自身不相容問題的研究進行詳細介紹。
1.2.1 獨立固定化催化劑 將化學-酶催化反應中的某一種催化劑采用固定化技術固定于載體中,將其限制在載體內部。同時借助載體上的孔阻止另一種催化劑進入載體內部,使二者在空間上無法接觸。載體的選擇對催化劑的性能有著至關重要的影響。一般來說,載體的選取需要遵循以下幾個原則:(1)比表面積大,有足夠的空間容納催化劑;(2)穩定性高,不僅要具有很高的機械穩定性以免材料塌陷,還要具有很好的化學穩定性避免在反應過程中引發副反應;(3)制備過程簡單;(4)環保無毒;(5)成本低廉。
介孔硅基材料[45-46]憑借著高穩定性、低毒性和高比表面積等優勢在眾多材料中脫穎而出。Inagaki等[47]在研究中發現Rh 配合物與牛血清白蛋白(BSA)的直接接觸會嚴重影響Rh 的活性。于是他們將Rh 配合物固定于聯吡啶基周期性介孔有機二氧化硅(BPy-PMO)中,介孔的存在使蛋白質無法進入BPy-PMO 內部與Rh 接觸。隨后將Rh@PMO 與馬肝醇脫氫酶(HLADH)結合,用于合成(S)-4-苯基-2-丁酮。由于避免了催化劑的失活,產品具有較高的轉化率和對映選擇性,并且Rh@PMO在2-環己烯-1-酮轉移加氫反應中表現出較高的催化活性和對酶的耐受性。類似地,Akai 等[48]將制備的氧化釩催化劑固定在孔徑為3 nm 的介孔二氧化硅(MPS)中得到V-MPS。MPS 有效地將化學催化位點與酶催化位點分隔開,使兩種催化劑能夠在同一介質中相容。
將有機金屬催化劑包埋在蛋白質支架中制備人工金屬酶被認為是解決催化劑接觸失活的有效方法。金屬催化劑被限制在宿主蛋白內部,有效地避免了與外部其他酶的接觸。Ward 等[49]將金屬復合物[Cp*Ir(Biot-p-L)Cl]包埋到鏈霉親和素(Sav)中制備出人工轉移氫化酶(ATHase)。ATHase 保留了貴金屬的催化活性,并且與天然酶具有良好的相容性。為了證明該方法的通用性,該課題組成功地將ATHase與三種不同的天然酶結合,設計了一系列級聯反應。該課題組利用這一策略還制備出了具有原位再生NADH 能力[50]和依賴NAD(P)H[51]的人工金屬酶,并應用于不同的級聯反應。
然而單獨固定某一類催化劑存在一定的隱患,因為固定后的催化劑難免會發生泄漏,泄漏的催化劑同樣存在使另一種催化劑失活的可能。將兩類催化劑分別固定于不同載體上,不僅可同時提升兩種催化劑的穩定性,還可借助載體對催化劑的保護作用消除催化劑泄漏所帶來的隱患。理論上,對于分隔催化劑活性中心的效果而言,將兩種催化劑分別固定在不同載體上是更優的選擇。
金 屬 有 機 骨 架(metal-organic frameworks,MOFs)以其可調的孔道結構、高比表面積和大孔隙率等優勢近年來受到了研究者們的廣泛關注[52-53]。Li 等[54]將Pd 固 定 于MIL-101 中,同 時 還 制 備 了CalB-CLEAs 交聯酶聚集體,利用二者開發了一種高效的化學-酶催化體系用于1-苯乙胺的動態動力學拆分。該過程采用微波輻射技術不僅加快了外消旋速率,還降低了反應溫度。固定化后的催化劑具有超高的轉化率和對映選擇性,而且重復使用9次后仍可維持很高的活性。Groger 等[55]將化學和生物催化劑分別固定于不同的高吸水性樹脂上,用于合成1,3-二醇。
1.2.2 分隔式集成催化劑 集成催化劑一般分為兩類,一類需要載體作為媒介,將酶與金屬共同固定于載體上;另一類則無須載體直接通過生物結合的方式將金屬連接在酶表面的氨基酸殘基上。無論是生物結合法還是簡單無差別的共固定方法都無法滿足催化劑空間分隔的目的,因為在制備過程中不同催化劑就已經發生接觸失活。將不同的催化劑有差別地固定在同一載體的不同位置上制備分隔式集成催化劑,才能有效地杜絕催化劑發生接觸失活。分隔式集成催化劑的另一大優勢是在避免催化劑活性中心接觸的前提下,大大縮短了活性中心的距離,從而提高中間產物的局部濃度,加快反應速率并且使反應平衡向產物的方向移動。制備具有空間分隔催化劑能力的先進納米材料是該策略的前提條件。在過去的幾十年中,納米材料領域的迅速發展使得研究者可以制備出所需的載體并對其可進行精準調控。制備分隔式集成催化劑一般需要將固定化酶和固定化金屬的方法結合起來,前者一般有物理吸附、共價交聯和包埋法;后者有共價交聯和前體原位生長法。
Jiang 等[56]制備了一種以PtPd 雙金屬為核、介孔多巴胺為殼的核殼結構納米顆粒。首先,將Pt和Pd的前體同F127 充分地溶解于水中。然后向混合液中滴加多巴胺溶液,多巴胺有足夠的還原力將前體還原成金屬顆粒同時自聚形成聚多巴胺(PDA)殼層包裹在金屬外表面。去除模板后得到PdPt@PDA 納米顆粒。隨后,采用共價結合法將酶固定于PDA 外表面得到分隔式集成催化劑,如圖5 所示。多巴胺殼層有效地將酶與雙金屬核分隔開,避免了催化劑之間的接觸失活現象。介孔結構顯著地提高了傳質和催化劑的利用率,進而加強了級聯反應的協同催化能力。該集成催化劑不僅可以實現一系列伯胺的動態動力學拆分(收率高達99%,光學純度高達98%),還可以將有機磷農藥級聯降解為低毒的對氨基苯酚。Prasad 等[57]制備了類似的金屬核-殼-酶結構的分隔式集成催化劑。以Au納米顆粒為核、氧化硅為殼層制備Au@mSiO2,將葡萄糖苷酶接枝到載體的外表面用于化學-酶級聯催化反應,同樣取得了不錯的效果。
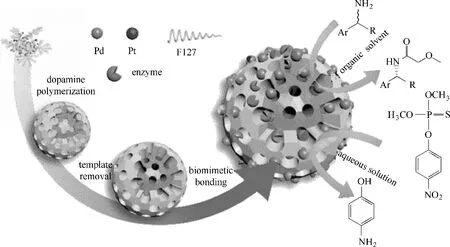
圖5 PdPt@PDA@CalB的制備過程[56]Fig.5 Formation process of PdPt@PDA@CalB [56]
Wu 等[58]將Pd 納米粒子(NPs)和CalB 分步負載到功能化介孔氧化硅納米顆粒不同位置。首先,利用前體原位生長法將Pd NPs 固定于MSN 上得到Pd-MSN。為了提高集成催化劑在有機溶劑中的溶解性,改變顆粒的親疏水性,采用長鏈烷烴修飾載體,進而得到疏水性的Pd-mMSN。最后通過疏水相互作用將酶固定于載體上得到CalB@Pd@mMSN。該方法不僅為催化劑的循環利用提供了多相載體,而且還避免了催化劑之間的相互失活。通過表面烷基化修飾后,雙功能催化劑可以分散在8 種有機溶劑中,因此所制備的催化劑在甲苯中合成己酸芐酯具有優異的催化性能。Qi 等[59]將Pd 原位生長在MOFs 的孔道結構內,隨后通過與月桂酸進行配體交換改變其在有機溶劑的分散性,最后將CalB 等酶固定于不同于Pd 的其他位置上。所制備的催化劑在苯甲醛和己酸乙酯合成己酸芐酯的過程中表現出優異的催化活性。
除了單一孔徑材料之外,多級孔材料很好地契合了空間分隔催化劑的需求。Yang 等[60]構建了一種多級孔硅基蛋黃殼@殼結構用于空間分隔Pd NPs與CalB。首先,采用前體原位還原法將Pd NPs 固定于氨基修飾的介孔氧化硅納米微球上得到Pd/NH2-MSN。隨后采用有機硅輔助刻蝕技術制備蛋黃殼結構的Pd/NH2-MSN@BTME。通過雙向分層方法,微球被具有大孔的介孔氧化硅殼包裹。最后采用吸附法將CalB 固定于大孔介孔孔道中,而不會進入固定Pd NPs 的小孔中。該結構設計不僅實現了兩種催化功能在一個納米反應器中的固定化,而且還實現了不同催化活性位點的空間分隔。
細胞膜作為天然的“載體”,也為制備分隔式集成催化劑提供了一種可行的策略。細胞膜不僅可以為酶提供最佳的內部環境避免其受到包括化學催化劑在內的外界影響,而且利用整個細胞進行催化直接省略了材料制備、分離純化酶和固定化酶的過程,僅需將化學催化劑固定在細胞表面即可。Lloyd 等[61]通過生物還原的方法將Pd NPs 固定在可過表達重組單胺氧化酶(MAO-N-D5)的大腸桿菌表面,用于合成對映體純的手性胺類化合物。該研究首次報道了全細胞生物金屬集成催化劑用于一鍋多步反應,提供了一種可擴展和高度靈活的平臺技術。受到該研究的啟發,研究者們基于全細胞開發了一系列集成催化劑用于一鍋化學-酶級聯反應[62-65]。
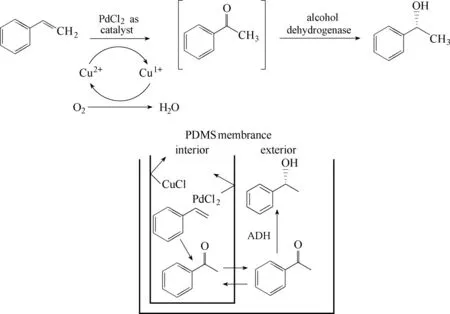
圖6 利用PDMS套管分隔催化劑活性位點實現Wacker氧化和酶還原反應的級聯反應[66]Fig.6 Site-isolation of catalysts using a PDMS thimble for the combination of a Wacker oxidation and an enzymatic reduction[66]
1.2.3 膜分隔(membrane-base separation) 膜分隔技術是將兩種催化劑分別限制在膜的兩側,選取合適的膜材料限制催化劑的通過,達到催化劑空間分隔的目的。Groger 等[66]在結合CuCl2/PdCl2催化的苯乙烯Wacker 反應和ADH 催化的還原反應時發現銅鹽對ADH 有著強烈的抑制作用。聚二甲基硅氧烷(PDMS)膜可以很好地將化學催化與酶催化分隔開,從而避免了化學催化劑對酶的損害。如圖6 所示,在PDMS 套管內部進行Wacker 氧化,中間產物在膜內的濃度不斷升高,逐漸向膜外擴散,而化學催化劑不會擴散到膜外而是被限制在膜內。擴散到膜外的中間產物被ADH 還原,從而使反應平衡向產物的方向移動,并且ADH 同樣限制在膜外。通過該策略在水相中將苯乙烯成功地轉化為1-苯乙醇(ee98%~99%)。
Latham 等[18]同樣采用PDMS 膜分隔技術將催化劑的活性位點有效地分離,并且將酶制備成交聯酶聚集體增大酶的體積,使酶能夠被嚴格地限制在膜的一側。隨后PDMS 膜分隔技術還被用于更多的化學-酶催化體系[67-68]乃至多酶體系[69],證實了該方法的通用性。
1.3 小結
本節介紹了時間和空間分隔等手段解決一鍋化學-酶級聯反應中常見的催化條件和催化劑本身的不相容問題,部分實例見表1。催化條件主要包括溫度、pH、底物濃度和溶劑的不相容。溫度與pH 的不相容通常采用連續反應的方式來解決;底物濃度對催化劑的影響需添加溶劑進行稀釋;而溶劑帶來的問題可借助兩相體系和非常規溶劑加以規避。催化劑本身的不相容需要采用制備分隔式集成催化劑和膜分隔的方法將催化劑的活性中心在空間上分隔。這些方法是互補的,在解決實際問題時可選取兩種乃至更多的方法以達到最優的效果。

表1 克服化學-酶催化不相容性的實例Table 1 Examples of overcoming chemoenzymatic incompatibility
2 化學-酶級聯催化的應用
2.1 動態動力學拆分
胺類和醇類等化合物的動態動力學拆分(DKR)是化學-酶催化反應應用最為廣泛的領域之一[70-71]。它由酶催化動力學拆分(KR)和金屬催化原位消旋化反應組成,理論產率為100%,克服了傳統的動力學拆分產率最高只有50%的局限。對映體純胺類和醇類化合物是重要的合成原料,廣泛應用于農藥、食品添加劑、香料和醫藥等化工產品的生產。
脂肪酶具有優異的對映選擇性、通用性和良好的疏水活性,常被用于手性醇和胺的動力學拆分。Jiang 等[56]制備的雙金屬- 酶集成催化劑PdPt@PDA@CalB 完成了一系列胺衍生物的DKR 反應,并取到了很高的收率與對映選擇性,如圖7 所示。同樣地,B?ckvall 等[72]將Pd 和CalB 共固定于硅基泡沫中用于對伯胺的DKR 反應,與游離的催化劑相比,固定后的催化劑反應效率和轉化數更高。Ge等[73]還對比了不同種類Pd-CalB 催化劑用于動態動力學拆分1-苯乙胺的催化能力。
與胺類化合物相比,催化醇類DKR 反應的催化劑種類更為廣泛,如V、Al、Ir 等金屬配合物,最為常用的是Ru配合物。B?ckvall 等[74]對此進行了大量研究,Ru 基配合物與不同類型的酶結合用于脂肪醇、丙烯醇、氯乙醇、二醇、高烯丙醇和N-雜環1,2-氨基醇的DKR過程。Nolan等[75]報道新型陽離子Ru配合物用于催化仲醇的消旋化(該過程避免了強堿的使用),與脂肪酶結合用于一系列伯醇的拆分。此外,Akai 等發現釩絡合物[VO(OSiPh3)3]與各種脂肪酶結合,可用于鏈狀[76]和環狀[77]烯丙基伯醇的DKR 反應。隨后他們還制備了一種新型的固定化金屬催化劑(V-MPD)[48],用于苯甲醇、雜環芳烴和炔丙醇的DKR反應,如圖7所示。
除了醇類和胺類的DKR 外,脂肪酶還與堿催化劑或過渡金屬催化劑偶聯用于合成更復雜的化合物。Ramstrom等[78]通過堿催化的半硫代縮醛形成反應和CalB 催化的分子內酯化反應合成了一系列手性1,3-硫氧雜五元環化合物,最終產物具有良好的轉化率和對映體純度,如圖7 所示。該課題組將CalB 與一個動態的多米諾酮加成環化反應進行耦合[79],合成了一系列新的摻雜N、O和S的六元雜環。
2.2 藥物中間體的不對稱合成
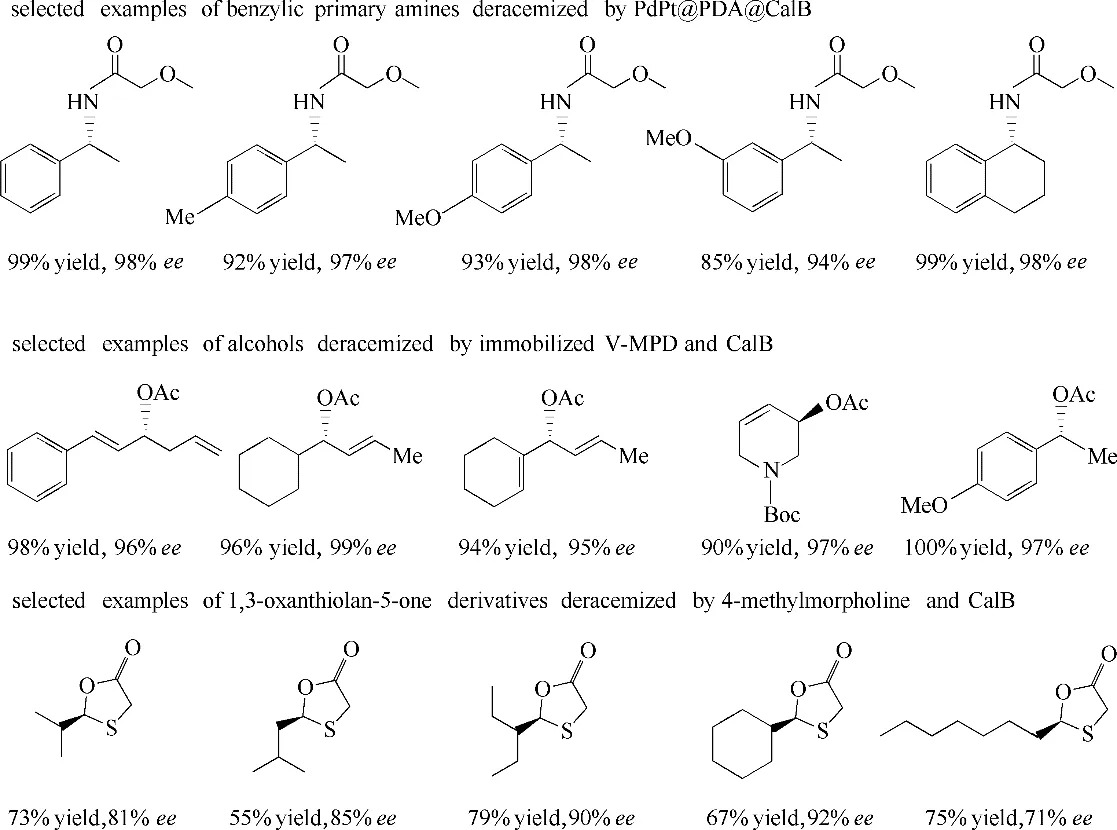
圖7 PdPt@PDA[56],V-MPD與CalB[48]和4-甲基嗎啉與CalB[78]動態動力學拆分的產物譜Fig.7 Scope of DKR systems involving PdPt@PDA[56],V-MPD and CalB[48],and 4-methylmorpholine and CalB[78]
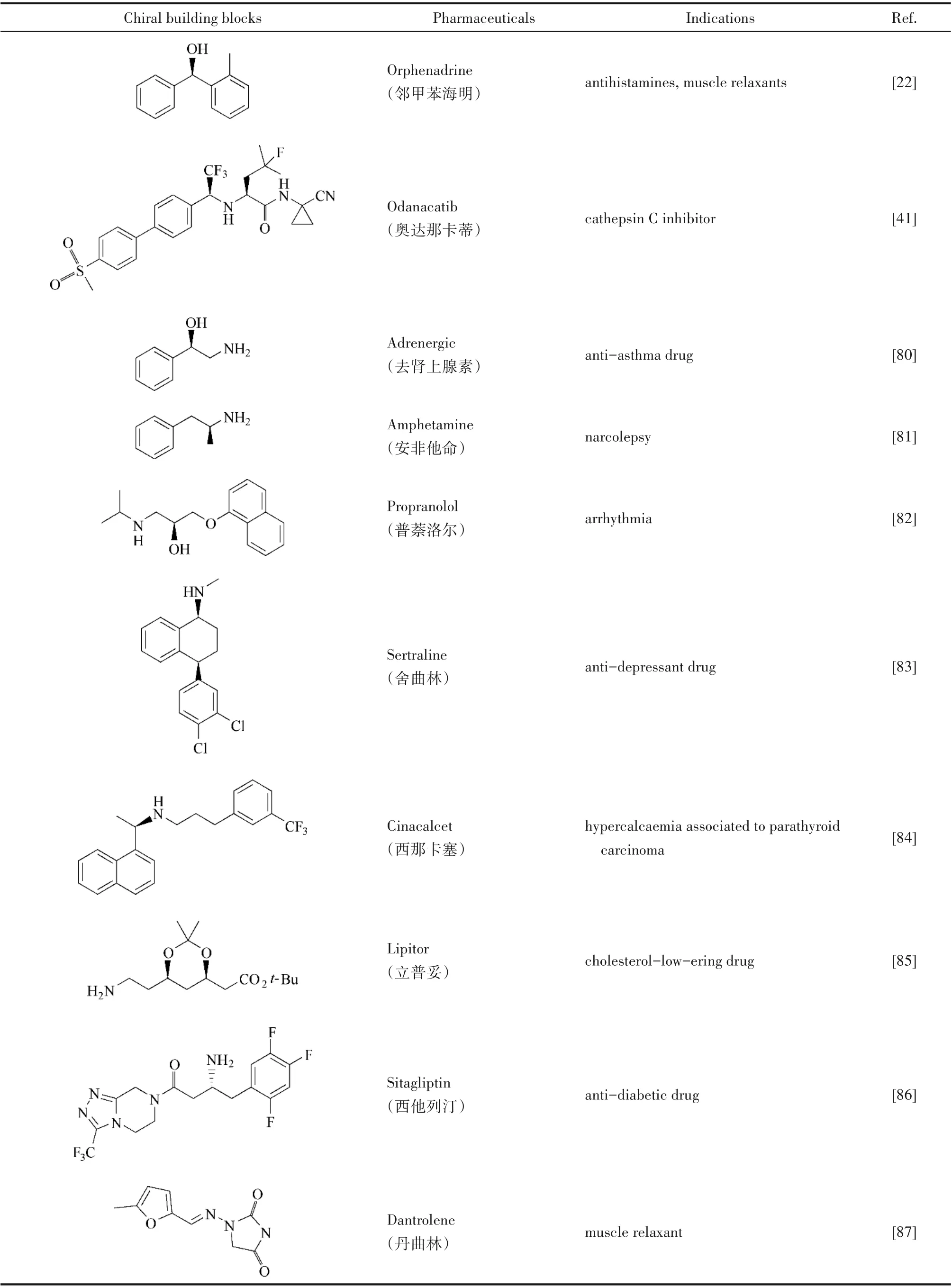
表2 化學-酶催化制備的手性藥物與中間體Table 2 Chemoenzymatic catalyzed preparation of chiral drugs and intermediates
目前市售的藥物有一半以上是手性化合物,化學-酶級聯催化在手性藥物中間體的合成領域也得到了廣泛應用。化學-酶催化合成藥物的典型例子包括西他列汀、辛古拉和立普妥的制備,其中立普妥是迄今為止最為暢銷的藥物之一。Garg等[22]通過將Ni 催化的Suzuki 交叉偶聯反應和KRED 進行偶聯,實現了在一鍋水介質中將胺類化合物轉化成手性醇,結合后續的烷基化反應成功制備了抗阻胺藥物奧芬那君。整個反應均在水相中進行,合成的產物不僅形成了新的C—C 鍵,而且還將其中一個參與反應的C 原子變為手性碳,省略了復雜的逆合成步驟。
Hollmann 等[80]將ADH 催化還原反應和Pd 催化的氫化反應結合,由此制備的2-氨基-1-芳基乙醇衍生物是合成抗炎、抗病毒或抗腫瘤藥物的重要中間體。利用該化學-酶催化成功合成的藥物活性成分(S)-替巴胺是一種天然的苯甲酰胺衍生物,具有抗病毒(HIV)活性,進一步突出了該反應的合成價值。Gotor-Fernández 等[81]將金屬催化的烯丙基苯Wacker-Tsuji 氧化反應與ω-TA 催化的生物還原反應相結合,在水介質中獲得了9 種具有光學活性的1-芳基-2-胺衍生物,其轉化率高達92%,對映體過量高達99%。該類化合物是合成苯丙胺類手性藥物的中間體。Xu 等[82]以環氧水解酶催化消旋的α-萘基縮水甘油醚拆分為關鍵步驟,通過后續的化學轉化合成了具有生物活性的普萘洛爾(一種典型的β 受體阻滯劑)。該藥物可用于治療心絞痛和心律不齊等疾病。
Berglund 等[83]開發出一種制備抗抑郁藥舍曲林的化學-酶級聯催化方法。KRED 催化外消旋四氫萘酮的生物還原,以出色的光學純度和非對映選擇性(ee>99%,dr>99∶1)合成(S,S)-醇。隨后利用次氯酸鈉作為氧化劑和2-氮雜金剛烷氮氧化物(AZADO)作為有機催化劑,將生成的(S,S)-醇氧化為對映體(S)-酮,得到舍曲林的重要手性前體。該課題組[84]通過化學-酶法利用ω-TA和KRED設計了兩種合成路徑,均成功合成了能激活甲狀旁腺中的鈣受體,從而降低甲狀旁腺素(PTH)分泌的西那卡塞。通過化學-酶級聯催化合成的手性藥物和手性藥物中間體還有很多,表2 對其中的一些典型案例進行了總結,并介紹了藥物的適應癥。
3 結 論
本文重點介紹了克服化學-酶催化過程中催化條件和催化劑不相容的策略,并對化學-酶級聯催化反應的應用進行了詳細匯總。采用時間分隔的連續反應模式、多相催化和非常規介質解決催化條件的不相容性;制備獨立固定化催化劑、分隔式集成催化劑和借助膜分隔技術在空間上分離催化劑活性位點,通過避免催化劑接觸失活解決催化劑之間的不相容性。這些方法充當了銜接化學催化與酶催化之間的橋梁。盡管如此,發展更為簡單通用的方法仍然十分迫切,這對級聯催化體系的發展起著關鍵性作用。今后,借助酶定向進化等技術對酶進行強化,使酶具有更強的抗逆性和更廣的反應性,可以大大增加酶與化學催化劑結合的機會,甚至使酶完全替代化學催化劑,使多酶體系取代化學-酶催化體系。另一方面,化學催化劑也將不斷發展,如在水相中的穩定性更好,反應條件更溫和,選擇性更高等。目前用于化學-酶級聯催化的化學催化劑大多數為貴金屬,例如用于動態動力學拆分的Ru 和Pd,在實際應用中開發廉價高效且綠色環保的催化劑是今后的工作重點。蛋白質工程、材料科學以及合成生物學等不同研究領域的發展和交叉融合將進一步推動化學催化與生物催化的結合,級聯催化的反應類型將不斷增加,實用性會更強,而且將越來越多地應用于工業化生產。
猜你喜歡
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
科技知識動漫(2016年10期)2016-10-18 20:35:00
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
新高考·高一物理(2014年1期)2014-09-18 01:26:07
