基于3D-QSAR和分子對接設計新型缺氧誘導因子脯氨酸羥化酶結構域蛋白1抑制劑
2020-12-24 07:52:52何華玉何清秀林治華
重慶理工大學學報(自然科學) 2020年11期
關鍵詞:模型
儲 涵,何華玉,何清秀,王 娟,林治華
缺氧誘導因子(HIFs)是由氧氣敏感的α亞基(HIF-1α、HIF-2α或 HIF-3α)和組成性表達的 β亞基(HIF-1β、HIF-2β或 HIF-3β)組成的異源二聚體轉錄因子[1]。在常氧環境中,HIF-α的羥基化和隨后的蛋白酶體降解能夠通過泛素連接酶系統對HIFs進行負調控[2-3]。同時,HIFs也是有限氧環境中氧穩態的主要調節器[4]。有研究表明:HIFs的穩定性主要由HIF-α的翻譯后脯氨酰羥基化來調節,該反應由缺氧誘導因子脯氨酸羥化酶結構域蛋白(PHDs)催化[5-6]。PHDs是一組在多細胞動物中充當氧氣感受器的酶,同時PHDs作為一種非血紅素鐵(II)依賴的酶,有3種催化活性亞型(PHD-1,PHD-2和 PHD-3)[7-8]。其中,PHD-1主要存在于睪丸中,但也存在于腦、腎、心臟和肝臟中,PHD-2存在于大多數組織中,PHD-3只存在于心臟中[9-10]。這3種異構體在其C端催化結構域具有很高的序列同源性,但在其N端沒有序列同源性[11]。
目前,在諸多臨床環境中,包括炎癥性腸病,缺血再灌注損傷和神經退變過程中,干擾PHDHIF軸成為了一種重要的治療方式[12-17]。常規的干擾方式(如Fe2+隔離劑等),由于缺乏特異性,在使用過程中存在很強的副作用[18-19]。1,2,4-三唑-[1,5-a]吡啶類化合物作為一類新型特異性PHD-1抑制劑,通過靶向 PHD-1干擾 PHD-HIF軸,避免了非特異抑制劑的副作用,也大大增強了療效。在本研究中,課題組收集了44個1,2,4-三唑-[1,5-a]吡啶類化合物,通過比較分子場分析(CoMFA)和比較分子相似性指數分析(CoMSIA),獲得了可靠的3D-QSAR模型。隨后,通過分子對接進一步分析了PHD-1與該類化合物的結合模式。最后,基于上述結果,設計了8種新型1,2,4-三唑-[1,5-a]吡啶類 PHD-1抑制劑,并確定可用于進一步研究的先導化合物。
1 材料與方法
1.1 數據選取
首先,根據文獻收集了 44個 1,2,4-三唑-[1,5-a]吡啶類化合物[20],將它們隨機分為訓練(36種化合物)和測試數據集(8種化合物,用符號“*”表示)。這些化合物的體外生物活性值以IC50表示,并轉換為相應的 pIC50(pIC50=-log IC50)。化合物的相關屬性見表1。
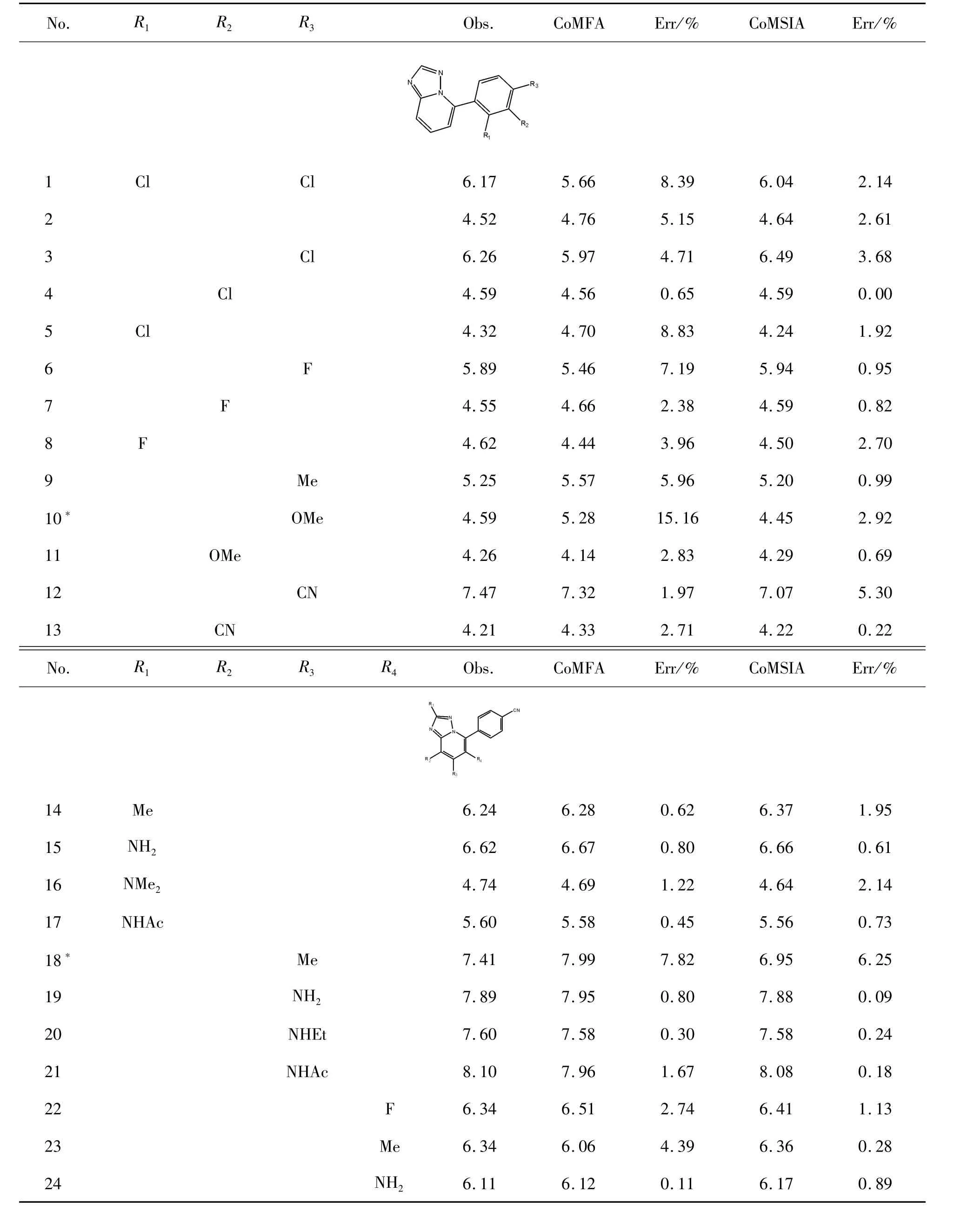
表1 化合物的實驗和預測活性(pIC50)值
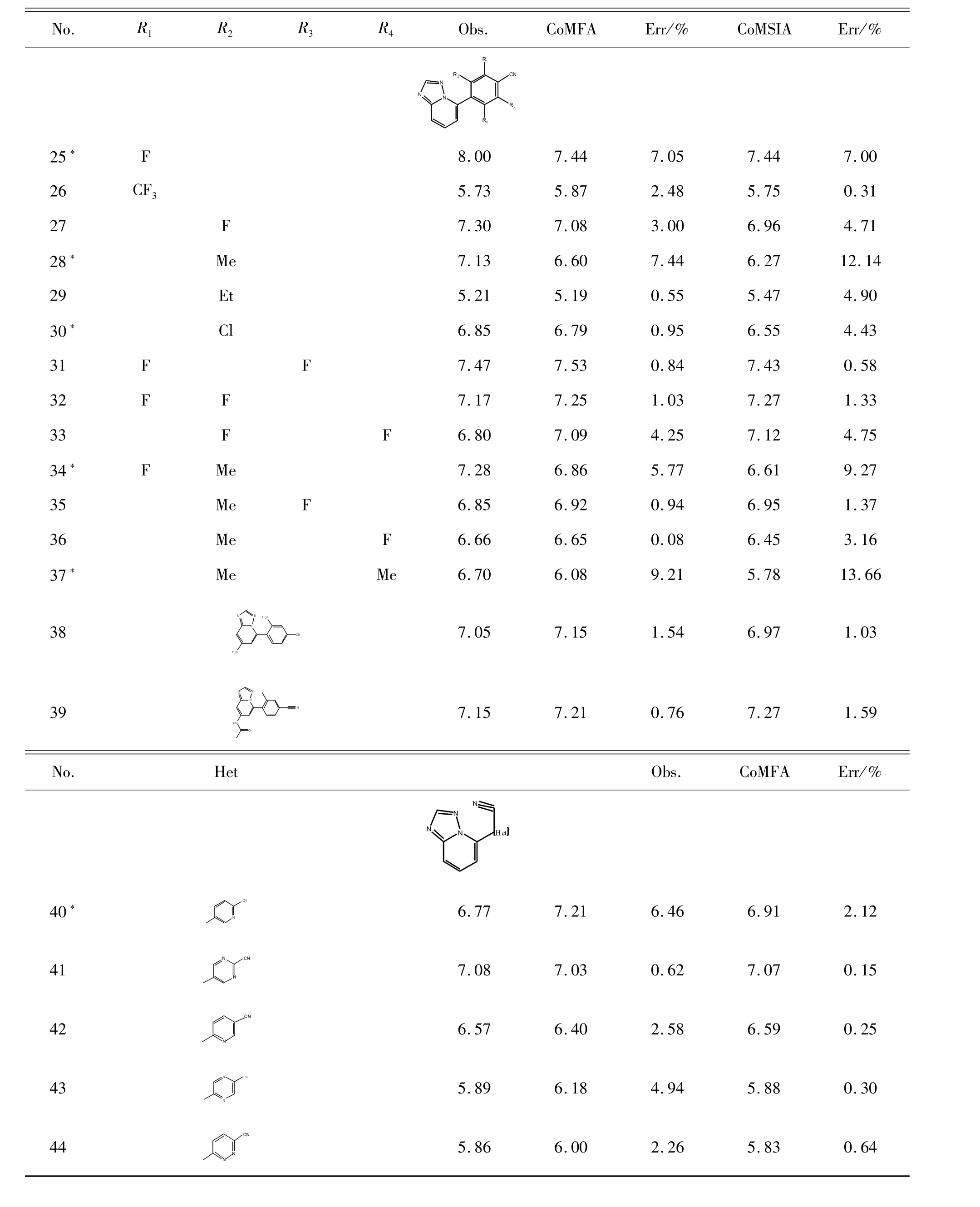
續表(表1)
1.2 結構優化和疊合
利用sybyl 2.0構建這44個化合物,然后給每個化合物附加Gasteiger-Huckel電荷和Tripos力場。采用Powll能量梯度法對化合物的結構進行優化,最大優化上限10 000次,收斂標準為0.005 kJ/mol,其余參數默認[21]。所有處理過后的化合物構象作為活性構象用于下一步的研究,同時化合物21(如圖1,圖中加粗部分表示公共骨架)具有最高的抑制活性,可作為疊合模板。選擇適當的公共骨架后,所有化合物的疊合如圖2所示。
1.3 構建3D-QSAR模型
CoMFA通過分析一組相似化合物周圍分子場的差異,得到這些分子與受體之間的非間相互作用的特性,從而預測其生物活性。CoMFA的靜電場和立體場分別采用Coalomb和Lennard-Jones勢函數,而CoMSIA還引入了高斯距離函數,通過疏水,氫鍵受體和供體場計算探針與分子的每個原子之間的相似性指數[22-23]。通過留一法(LOO)交叉驗證程序計算了相關模型的最佳組件數(n)和最高交叉驗證相關系數(q2)。常規的多重相關系數(r2),估計的標準誤差(SEE)和 Fisher檢驗(F)值是通過非交叉驗證的分析獲得的[24-25]。利用3D-QSAR模型的等勢圖研究分子結構對生物活性的影響,從而指導化合物結構的修飾以提高其活性。
1.4 分子對接
PHD-1(PDB∶5v1b)晶體結構來源于蛋白數據庫(protein data bank,PDB),利用 Sybyl軟件對PHD-1晶體結構進行加氫、加電荷等處理,同時,去除晶體結構中的原配體小分子(8UY)。將原配體小分子范圍內0.5 nm的氨基酸殘基作為對接口袋區域,并以其為參考分子,其余參數為默認值[26]。基于上述參數,將所有化合物與其對接,如圖3,使用PyMOL軟件進行對接可視化。
2 結果與討論
2.1 3Q-QSAR模型
以36個 1,2,4-三唑-[1,5-a]吡啶類化合物為訓練集,CoMFA和CoMSIA建模后的計算結果顯示在表2中。CoMFA模型和CoMSIA模型的穩定性可以用交叉驗證的q2來表示。一般情況下,當q2大于0.3時,所構建的模型僅在5%水平上有統計學意義;當q2大于0.5時,模型具有顯著的統計學意義。
空間場和靜電場構建的CoMFA模型最佳,LOO的q2為0.712,表明該模型具有良好的穩定性和可預測性。此外,CoMFA模型的n值為7,r2為0.969,F值為123(認為可靠的F>100),SEE為0.222,說明該模型具有良好的統計意義。在CoMSIA模型中,空間場、靜電場、疏水場,氫鍵供體場和受體場的組合表現良好(q2=0.754,r2=0.985),同時其余參數也可證明(F=159,SEE=0.165)。3D-QSAR模型的訓練集與測試集的線性相關性分析如圖4所示。CoMFA與CoMSIA模型中訓練集與測試集的大部分化合物都位于或接近趨勢線附近,證明了化合物實際活性值與預測活性值(以pIC50表示)的擬合度較好。
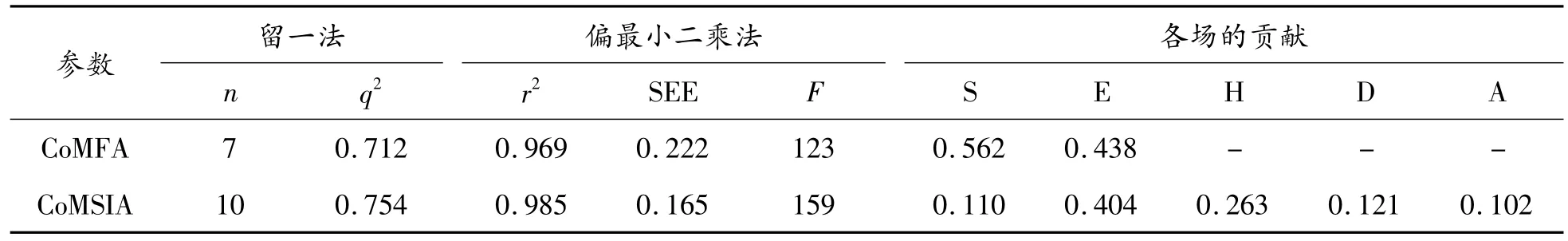
表2 3D-QSAR模型的計算結果
2.2 CoMFA和CoMSIA三維等勢圖分析
使用實際活性最高的的化合物21作為模板進行CoMFA與CoMSIA的三維等勢圖分析。如圖5所示,CoMFA立體場、COMSIA靜電場、疏水場和氫鍵受體場貢獻較高,可用于指導化合物結構修飾和改造。
圖5 (a)是 CoMFA的立體場輪廓圖。綠色(貢獻80%)和黃色(貢獻20%)區域的組合表明:在化合物的苯環連接的-CN處以體積較大的基團取代能夠增強活性。化合物10(pIC50值為4.59)在該處以較大體積的基團-OMe取代,相較于無取代基的化合物13(pIC50值為 4.21)活性更高。CoMSIA的靜電場等勢圖見圖5(b),藍色區域表示在此處增加負電性基團有利于提高化合物的活性;紅色區域表示在此處以正電性基團取代更好。這種結合表明:三唑環帶有負電荷的基團有利于活性。化合物19(pIC50值為7.89)的-NH2取代負電性更強,比-CH3修飾的化合物18(pIC50值為7.41)有更優異的活性。CoMSIA的疏水場等勢圖見圖5(c),白色區域表示在此處增加親水性基團有利于提高化合物的活性,黃色區域表示在此處以疏水性集團取代更好。因此,在化合物的苯環連接的-CN處以親脂性的基團取代能夠增強活性。CoMSIA的氫鍵受體場等勢圖顯示在圖5(d)中。紅色部分表示在此處減少氫鍵受體有利于活性的提升。因此,在苯環上的需要更多的非氫鍵供體。這些已通過系統中的某些化合物確認。在化合物26和27中,化合物27(pIC50值為7.30)以-H取代,提供氫鍵供體而非氫鍵受體,其活性明顯高于-CF3取代的化合物26(pIC50值為5.73)。
2.3 分子對接模型分析
為了探索PHD-1與配體的結合方式,利用SYBYL軟件的Surflex-Dock模塊進行分子對接,如圖6所示。在對接所有化合物前,將PHD-1共晶配體8UY重新對接到結合囊中來驗證所使用的方法和參數是否可靠可行。如圖7所示,8UY分子重新對接的構型與其共晶配體構型基本一致,兩種構型的RMSD為0.2?(<2.0?)。化合物中的三唑環上的N原子直接與金屬Fe結合,這種特征的相互作用與以往的研究是一致的。這也說明所生成的對接協議是可靠的,可用于后續的研究。
本研究選擇化合物21作為分析結合模式的模板,并用二維圖(圖8)和三維圖(圖6)進行展示。首先,化合物21三唑環上的氮原子與活性部位的Fe形成Fe-N鍵(2.0?)。與苯環相連的NH連接體與ASP238形成了一個距離為2.4?的強氫鍵。值得注意的是化合物另一端的氰基與Asn315也形成了一個強氫鍵(2.3?)。這種氫鍵的結合方式保證了其穩定性。此外,化合物21與PHD-1之間存在豐富的疏水相互作用。Tyr287與該化合物的苯環之間都存在Pi-Pi相互作用(3.3 ?)。此外,Ala369與該苯環之間也存在著一個疏水作用(5.9?)。另外,Ile311與三唑環之間還存在著一個弱疏水作用(3.9?)。豐富的疏水相互作用和氫鍵在配體與受體的結合中起著關鍵作用,它們可以使化合物與受體的結合更加穩定。
2.4 新型化合物的設計與評價
通過分析3D-QSAR的等勢圖和分子對接模型,以化合物21為先導化合物,確定該類化合物的修飾區域。通過在苯環附近引入氫鍵受體和體積較大的疏水性基團,在三唑環附近引入正電荷基團,本文共設計了 8個新的1,2,4-三唑-[1,5-a]吡啶類化合物。
對于這8個新設計的化合物,首先通過建立的3D-QSAR模型預測了其活性,其次將其與PHD-1對接并打分。化合物的結構,預測活性以及對接得分如表3所示。結果表明:所有新化合物抑制活性較高,且對接得分也較高,尤其是化合物21-g,其預測活性相較于化合物21有明顯的提升,同時它與PHD-1的對接得分也同樣有所提升。這表明,相較于其余修飾化合物的方式,在苯環附近引入氫鍵受體能夠很好地提高該類化合物與PHD-1結合的穩定性,從而大大提升1,2,4-三唑-[1,5-a]吡啶類化合物作為特異性PHD-1抑制劑的活性。
同時,由于化合物21-g具有出色的預測活性和對接評分,因此它可作為新型 1,2,4-三唑-[1,5-a]吡啶類PHD-1抑制劑的先導化合物,用作進一步的研究。
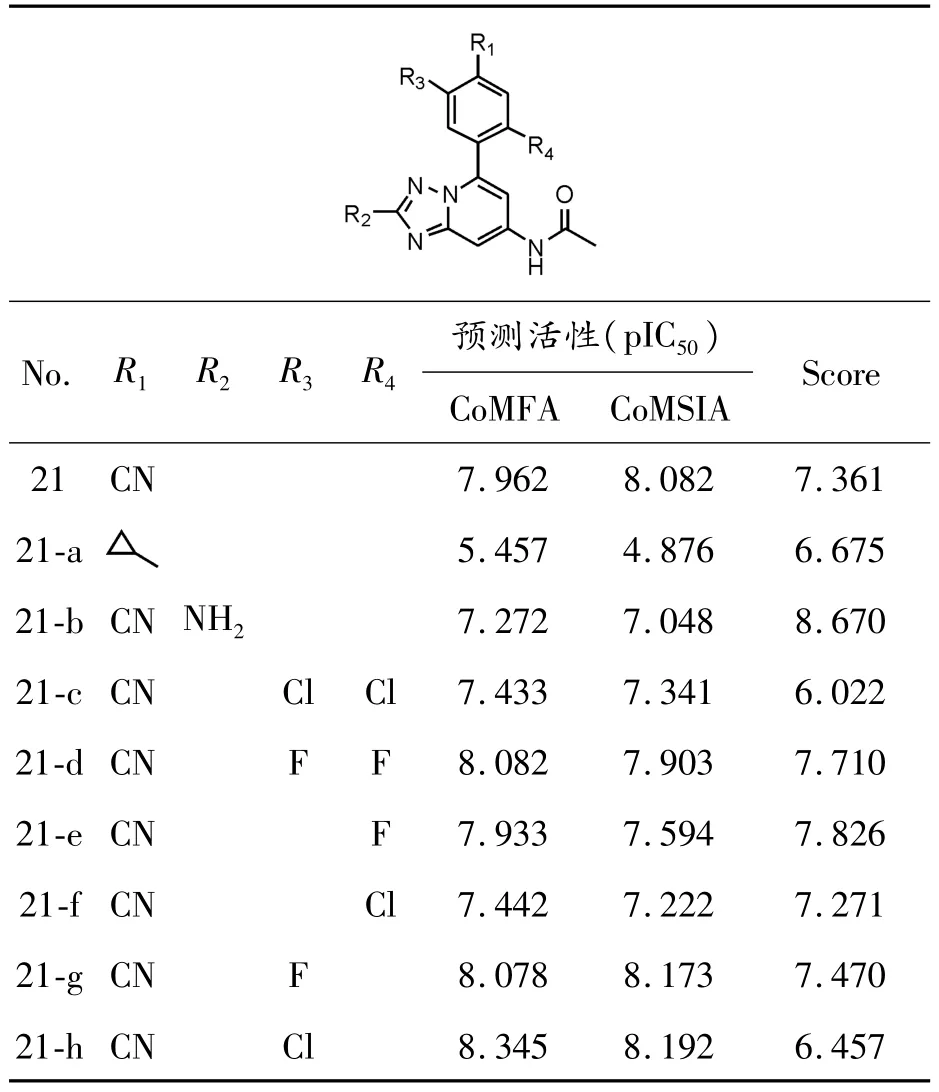
表3 新設計化合物的結構和預測活性
3 結論
本文中結合3D-QSAR和分子對接技術研究了1,2,4-三唑-[1,5-a]吡啶類 PHD-1抑制劑。所建立的3D-QSAR模型揭示了PHD-1抑制劑結構與活性關系的關鍵因素,這些關鍵因素可用于指導該類化合物的改造和設計。此外,分子對接技術被用于探索蛋白晶體活性位點中的關鍵氨基酸與配體分子的對接模式。通過分析空間場、疏水場、靜電場和氫鍵受體場的等勢圖,以及1,2,4-三唑-[1,5-a]吡啶類化合物與PHD-1的對接模型,設計了8種新的該類化合物。新型化合物評價結果表明:它們都具有良好的預測活性和較好的對接得分,尤其是化合物21-g,可作為先導化合物做進一步研究。這些結果為特異性PHD-1抑制劑的合理設計奠定了基礎。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
