蒲公英根提取物自微乳給藥系統的處方設計
2020-12-08 05:34:16王麗娜
中國獸醫雜志 2020年7期
王麗娜,賈 皓,于 同,王 丹,陳 爽,李 研
(1.吉林醫藥學院藥學院,吉林 吉林 132013;2.海南醫學院基礎醫學與生命科學院,海南 海口 570000)
自微乳給藥系統(Self-microemulsifying drug delivery system,SMEDDS)屬于各項同性系統,其外觀是澄清透明的,而且呈現穩定的熱力學及動力學狀態,可以在體內蠕動后或在體外加水攪拌狀態下自發地進行乳化作用,其粒徑為10~100 nm,是屬于水包油(O/W)型的微乳[1-5],在一定溫度的條件下長時間地放置或離心后都顯示比較穩定的狀態,而且一般粘性都較小,因此可以通過過濾進行滅菌,極其便于操作;因為其形成的微乳粒徑都比較小,所以可以作為納米級別的給藥系統,極大地增加了藥物的表面積,同時也有利于對藥物的吸收利用,還可以增大難溶性藥物的溶出度,從而使難溶性藥物在體內的生物利用度增大[6-8]。SMEDDS在口服后可以在胃腸道的蠕動下及胃腸道內水的參與下自發地形成水包油型微乳,能快速均勻地分布在胃腸道中,有利于增強胃腸道對藥物的吸收利用;除了可以減輕藥物對胃腸道的刺激外,還可以進一步增大體外的溶出度,同時還能使藥物在體內的吸收率增大;另外,還可使胃腸黏膜的清除作用有效地降低,除上述之外,還可以避免首過效應,對防止藥物被酶水解失活具有一定的作用,從而使藥物的生物利用度增大,另外也可使藥物在個體間的吸收差異降低,尤其適用于口服吸收效果差及生物利用度低的藥物[9-11]。
蒲公英是一種極其常見的植物,擁有多種多樣的別名,常被稱為婆婆丁,也被稱為黃花地丁等[12]。在全球范圍內分布廣泛,遍布于北半球溫帶至亞熱帶中部地區;我國也擁有非常多的蒲公英,大多分布在華北、東北、西南省區以及西北省區,另外在東南及華南省區也有少量分布。蒲公英有著清熱解毒及利尿散結的功效,因此常被作為中草藥和野生蔬菜使用,屬藥食同源類中藥材。藥理作用有抗腫瘤、廣譜抗菌、免疫調節等,被廣泛應用于臨床治療中,如常被用于急性乳腺炎、尿路感染等疾病的治療[13]。
蒲公英具有很多種的化學成分,主要包括咖啡酸、三萜類黃酮類、酚酸類、倍半萜內酯類、植物甾醇類和香豆素類等化合物[14]。目前已經開發出來的蒲公英藥物劑型包括片劑、注射劑等,但在自微乳方面還未有涉足,本試驗旨在更加深入地研究利用蒲公英,發揮其最大價值,同時為新劑型的開發提供基礎和理論指導。
1 材料與儀器
1.1 材料 咖啡酸標準品(批號:331-39-5),購自北京盛世康普化工技術研究院;咖啡酸,購自湖北鑫鳴泰化學有限公司;1,2-丙二醇、鹽酸、甲醇、乙醇,均購自上海凌峰化學試劑有限公司;薄荷油,購自黃山天目薄荷藥業有限公司;吐溫60、吐溫40、吐溫80、油酸乙酯、聚乙二醇400,均購自國藥集團化學試劑有限公司;其他試劑為分析純。
1.2 儀器 日本島津LC-20AT高效液相色譜儀;LC-20AT泵;LC-solution色譜工作站;ZRS-8G型溶出度測試儀,購自上海書培試驗設備有限公司;KQ-250DE型數控超聲波清洗器,購自昆山市超聲儀器有限公司;TG16MW型臺式高速離心機,購自湖南赫西儀器裝備有限公司;ZS90型粒度分析儀、恒溫磁力加熱攪拌器(IKA)、0.45 μm微孔濾膜,均購自天津市津騰實驗設備有限公司。
2 方法與結果
2.1 蒲公英根提取物干燥粉末的制備 將75%乙醇回流提取液少量多次加入到蒸發皿里,將蒸發皿放置在烘箱中,調節溫度為100 ℃,待蒸發至完全干燥不黏稠時,取出置于室溫下,待冷卻后用藥匙刮下干燥粉末,放置在研缽中研碎后過100目篩,避光干燥保存。
2.2 空白自微乳的處方篩選
2.2.1 藥物的溶解度測定 分別稱取過量且質量相同的蒲公英根提取物干燥粉末,加入到燒杯里,再分別取1 mL薄荷油、油酸乙酯、油酸加入到其中,在37 ℃恒溫水浴下加熱10 min,直到不再溶解,再分別測定咖啡酸、總黃酮的含量。
同法測定藥物在乳化劑吐溫40、吐溫60、吐溫80中的溶解度,通過觀察判斷可以得出,藥物在其中的溶解度順序為:吐溫80>吐溫60>吐溫40,同樣,藥物在助乳化劑即甘油、乙醇、PEG-400、1,2-丙二醇中的溶解度順序為:1,2-丙二醇>甘油>乙醇>PEG-400。根據藥物在各物質中的溶解度可以確定最終自微乳處方的油相為薄荷油,乳化劑為吐溫80,助乳化劑為1,2-丙二醇。見表1。
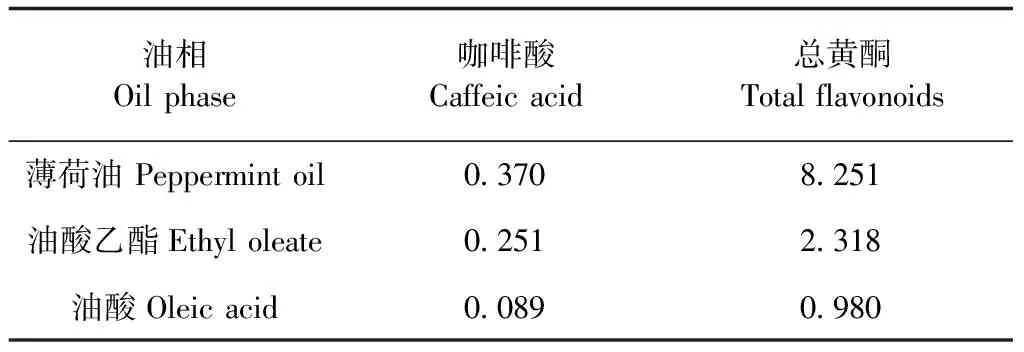
表1 藥物在不同油相中的溶解度
2.2.2 偽三元相圖的繪制
2.2.2.1 自微乳Km值的確定 設定固定油相與混合表面活性劑的質量比為1∶1,分別按1∶9、2∶8、3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1的質量比例精密稱取乳化劑與助乳化劑,放入到干燥潔凈的燒杯中,再加入油相,在(37±0.5)℃的恒溫水浴中、在轉速為500 r/min的條件下進行磁力攪拌,先將自微乳混合均勻,再將水逐滴加入其中。肉眼觀察其外觀變化,在某一刻能形成澄清透明,或帶有少許藍色乳光的溶液,此時即為形成微乳的臨界點,將該點確定為偽三元相圖中可形成微乳的區域點(不包含水相邊線),準確記錄加水量,計算形成微乳臨界點時各組分所占的質量分數[15],從而繪制偽三元相圖。相圖的3個頂點分別是油相、混合表面活性劑和水相,用Orign 8.0軟件繪制偽三元相圖,確定所成微乳區面積,篩選乳化劑和助乳化劑的最佳質量比,即Km值。偽三元相圖如圖1所示,當乳化劑與助乳化劑比例為5∶5時,即Km值為1時所形成微乳區域的面積最大。以偽三元相圖中形成微乳區域的面積和溶液外觀作為判斷標準,選擇最佳乳化劑、助乳化劑的比例,當Km=1時所形成的微乳澄清透明,并帶有藍色乳光,在放置48 h后不分層,其他比例的微乳均有明顯分層現象,當乳化劑∶助乳化劑為1∶9時進入凝膠區,呈透明黏稠液體,轉子停止轉動,不利于在體內自發乳化。見圖1。
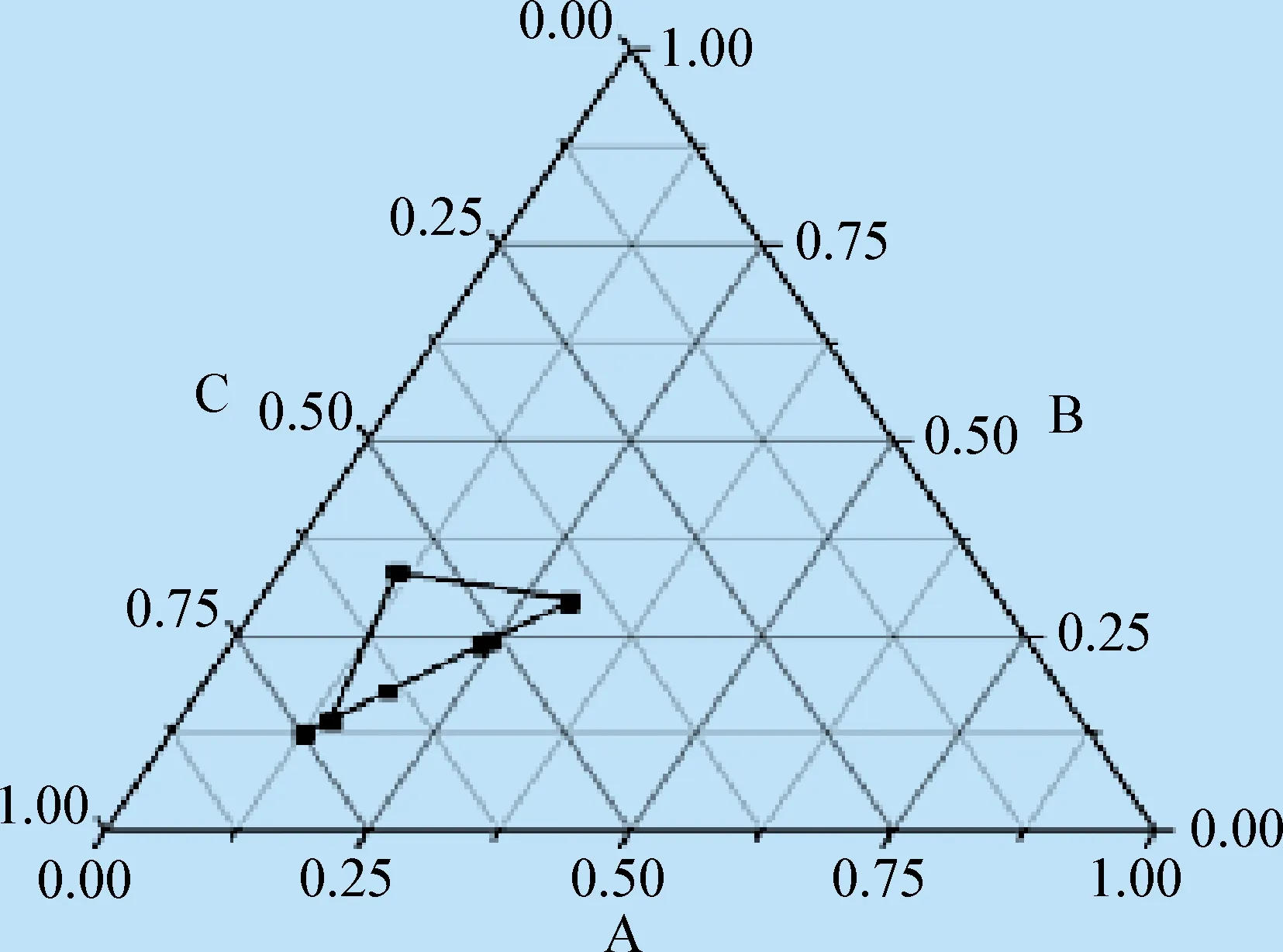
圖1 篩選自微乳給藥系統Km值的偽三元相圖
2.2.2.2 篩選油相與混合表面活性劑最優比例 固定乳化劑與助乳化劑Km值為1,改變油相所占比例,使其比例分別為10%、20%、30%、40%、50%、60%、70%,混合表面活性劑的比例隨之發生相應改變,即乳化劑與助乳化劑均分別為45%、40%、35%、30%、25%、20%、15%,按“2.2.2.1”項操作,篩選最優比例。其中當油相比例占10%時,加水攪拌后呈黃色油狀液體,無變化,不能形成微乳;當油相比例占20%時,加水攪拌后溶液澄清透明且有明顯藍色乳光出現。偽三元相圖中形成微乳區域面積表明當薄荷油比例為20%、吐溫80比例為40%、1,2-丙二醇比例為40%時為最優處方,外觀也呈明顯澄清透明且有藍色乳光的溶液。偽三元相圖見圖2。

圖2 篩選油相與混合表面活性劑最優比例
2.3 星點設計-效應面法優化處方
2.3.1 蒲公英根提取物自微乳的制備 分別按比例精密稱取油相、乳化劑和助乳化劑,再稱取過量蒲公英根干燥提取物加入其中,在60 ℃恒溫水浴中,轉速為500 r/min的條件下在磁力攪拌器上進行攪拌,以促進藥物在自微乳中的溶解,然后在37 ℃ 恒溫水浴中平衡10 min,12 000 r/min離心10 min 后,取上清液即得加藥自微乳。
2.3.2 蒲公英根提取物自微乳粒徑的測定 稱取蒲公英根提取物自微乳至燒杯中,再加入25倍(37±0.5)℃的蒸餾水,在轉速為500 r/min的條件下,保持水浴(37±0.5)℃,磁力攪拌均勻后,再用馬爾文激光粒度儀測定所形成自微乳的粒徑。
2.3.3 蒲公英根提取物自微乳總黃酮載藥量的測定 制備加藥自微乳,用1 mL移液管精密移取1 mL上清液加入到10 mL容量瓶里,再用濃度為75%的乙醇定容到刻度線處,搖晃均勻后,從其中取出1 mL溶液,進行總黃酮含量的測定。
2.3.4 星點設計-效應面法優化處方 在偽三元相圖篩選空白自微乳處方的基礎上,選擇影響自微乳性質較大的2個因素,將油相質量分數(X1)及Km(X2)作為考察因素,采用偽三元相圖法對Km值進行試驗,結果顯示,當Km=4 時,有較大面積凝膠區域出現,因此Km的范圍為1~3;自微乳體系的形成與油相質量分數所占比例有關,當油相所占比例越小,則SMEDDS越容易形成,故將油相質量分數的范圍設置在10%~40%。以咖啡酸載藥量、總黃酮載藥量和粒徑為評價指標,采用星點設計-效應面法對處方進行優化[16-17],因素水平如表2所示,試驗設計及結果如表3所示。
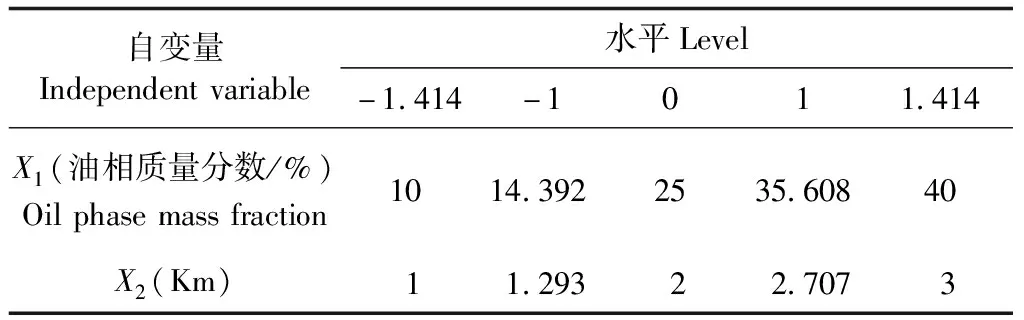
表2 星點設計因素水平
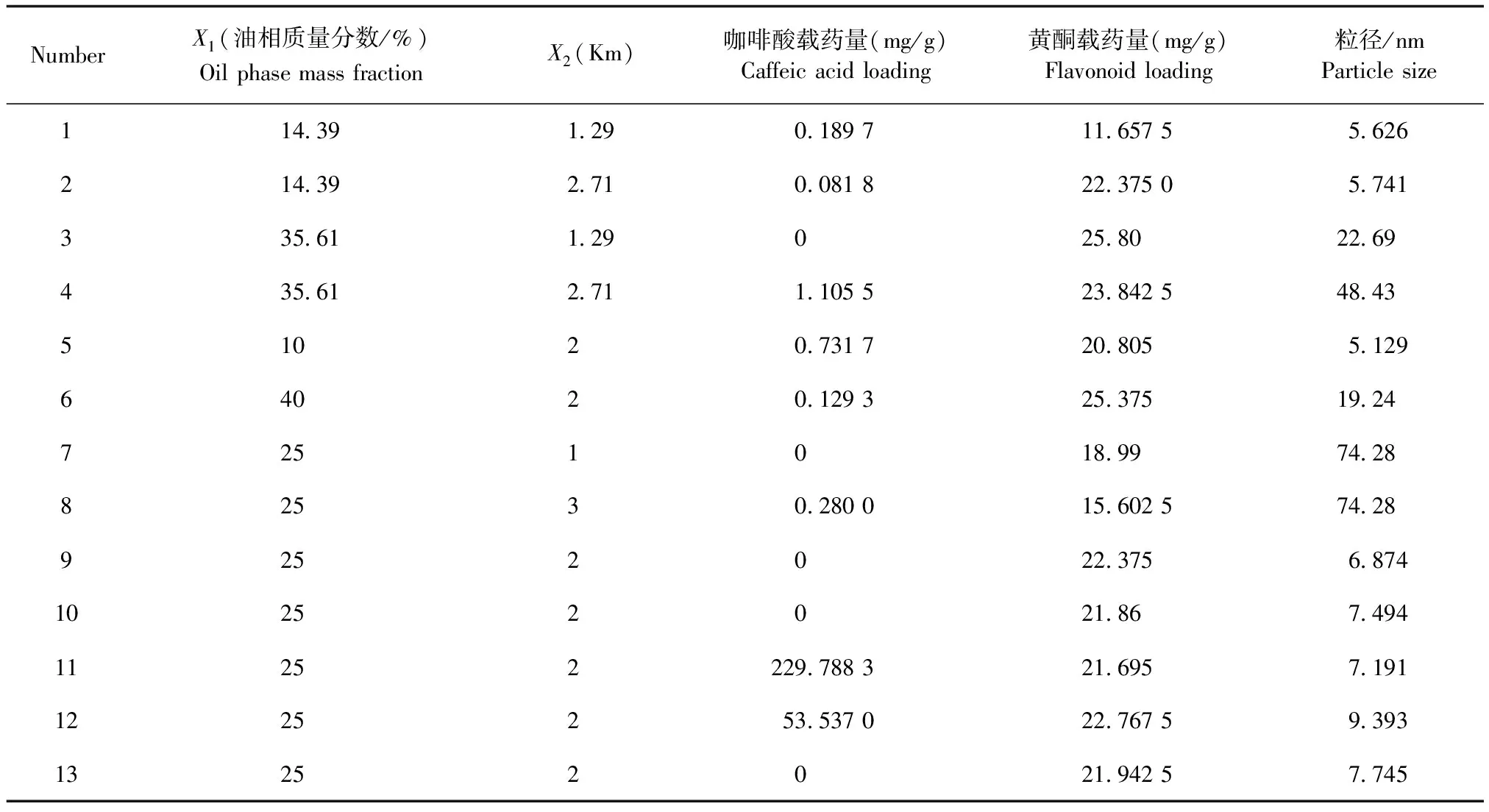
表3 星點設計及結果
2.3.4.1 模型擬合 星點設計試驗需要對各因素水平進行非線性擬合,即二項式擬合,因此運用Design Expert 8.0.6 軟件,做出二項式擬合的模型,其結果分別為:
黃酮載藥量(Y1)=-21.105 53+0.810 79A+291.145 65B-0.422 51AB-6.51A2-22.89B2(R=0.975);
咖啡酸載藥量(Y2)=137.238 69+0.672 89A+13.291 435B+107.2 491 235AB-29.489 5A2-46.365 85B2(R=0.949);
粒徑(Y3)=219.781 86+0.754 97A-237.791 47B+0.854 18AB-0.030 482A2+55.252 04B2(R=0.928)。
二項式擬合方程的相關系數分別是0.975、0.949和0.928,相關系數均達到較高值,該設計模型擬合效果良好,結果具有統計學意義,因此可以用該模型對蒲公英根提取物自微乳處方進行分析和預測[18]。
2.3.4.2 效應面優化 采用Design Expert 8.0.6 軟件,分別繪制2種不同影響因素對于3個響應值的三維曲線,黃酮載藥量的效應曲面如中插彩版圖3所示,咖啡酸載藥量的效應曲面如中插彩版圖4所示,粒徑的效應曲面如中插彩版圖5所示。根據方程和響應面圖確定蒲公英根提取物自微乳的最優處方為:油相比例為35.61%,Km=1.58;黃酮載藥量理論值為26.345 8 mg/g,咖啡酸載藥量理論值為21.987 9 mg/g,粒徑理論值為51.528 7 nm。
2.3.5 驗證試驗 根據軟件優化所得的最佳處方,制備蒲公英根提取物自微乳驗證試驗,并分別對黃酮、咖啡酸載藥量和粒徑進行測定,結果見表4。通過驗證試驗可以證明,最優處方粒徑和載藥量的真實值與預測值之間存在顯著性差異,說明星點設計-效應面法所建立的模型具有預測性良好的特點,可以較好地應用于蒲公英根提取物自微乳給藥系統的處方篩選優化。
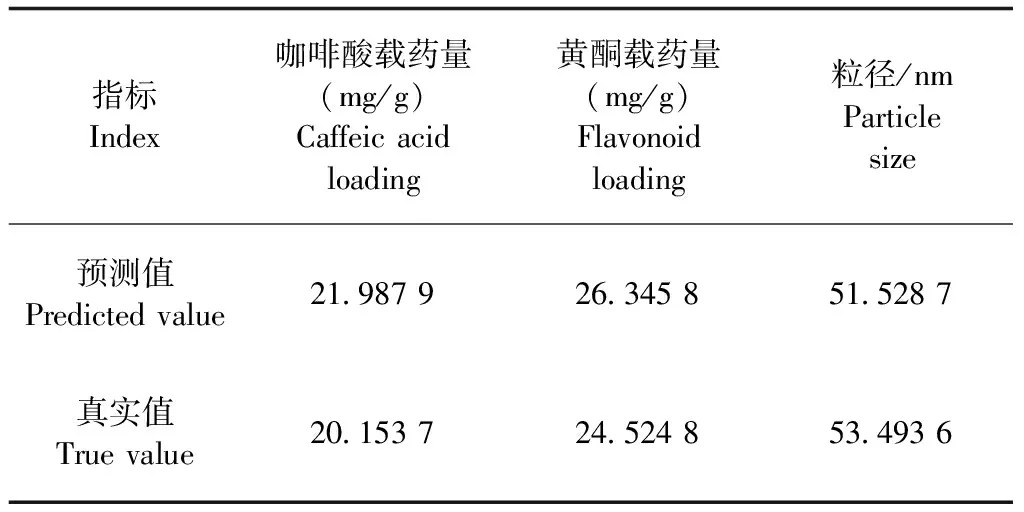
表4 星點設計-效應面優化法驗證
通過對多種不同條件下蒲公英根提取物進行黃酮含量測定,選擇黃酮含藥量最多的提取液,進一步進行處理,在制備蒲公英根自微乳時使用。油相的篩選中油酸在室溫下容易凝固,不符合自微乳油相的要求;乳化劑在篩選時同樣吐溫40和吐溫60在室溫條件下容易凝固失去流動性,不利于自微乳的制備,并且溶解度在其中也比在吐溫80中小。自微乳的粒徑與其在磁力攪拌器上的攪拌時間和轉速有關,故試驗過程中固定溫度、轉速和時間等外界條件。通過偽三元相圖先初步篩選空白自微乳的比例,在此基礎上再利用星點設計-效應面法進一步優化含藥自微乳處方,最終由星點設計建立的模型出最優處方。
3 討論
本試驗選擇的乳化劑吐溫80、吐溫60等為非離子型乳化劑,具有高親水親油平衡(HLB11-15)值,小腸上皮細胞膜的流動性可被有效改善,增加了細胞間的連接從而加速藥物的穿膜滲透,藥物的溶出度和生物利用度因此得到提高。處方中助乳化劑則選擇能與乳化劑相溶的低相對分子質量的醇類如PEG-400、1,2-丙二醇等,除了可以在其他相之外輔助溶解藥物,還可以提高膜的牢固性和界面膜的柔順性流動性,調節乳化劑的HLB 值,同時又可減少乳化劑的用量,從而降低自微乳的潛在毒性,有利于藥物最終的臨床應用。與傳統乳劑相比,蒲公英根自微乳溶液粒徑更小,能夠使得藥物更加分散,促使藥物更快、更有效地被胃腸道吸收,從而提高生物利用度。偽三元相圖用于空白自微乳的處方篩選,由于其不完全確定性及試驗過程中目測相變臨界點的人為因素,故可能存在誤差,且處方的覆蓋面可能不夠全面,因此先確定比例選擇各個相的大致范圍;再利用星點設計-效應面法進一步對加藥自微乳進行處方篩選,此種方法可有效針對試驗建立模型,在有效范圍內覆蓋大面積范圍進行試驗,相較偽三元相圖法篩選處方更加有效準確。
自微乳給藥系統可增加難溶性藥物的溶解度,口服后其在胃腸蠕動作用下快速乳化和高度分散,形成具有巨大表面積的區域,可有效提高藥物的吸收速率和程度;自微乳給藥系統中的表面活性劑成分可增加細胞膜的流動性,提高膜的通透性而促進跨膜吸收;自微乳給藥系統中脂質成分如脂肪酸等可提高藥物的淋巴轉運增加口服吸收,具有廣泛的應用前景。
