1例Miyoshi肌病的臨床、影像、病理及基因檢測報道
2020-11-01 10:25:30諶書云
康頤 2020年12期
諶書云
【摘要】目的:分析Miyoshi肌病的臨床影響及基因檢測情況。方法:回顧性分析1例Miyoshi肌病的臨床病例,并總結其臨床表現、影像學資料以及基因檢測報告。結果:患者足跟、足尖行走不能,行走跨閾步態;影像學檢查發現磁共振成像顯示下肢肌肉不僅存在萎縮脂肪化,還存在明顯水腫信號,主要以小腿中下部明顯。基因檢測結果發現二代測序顯示為dysferlin基因的復合雜合突變。結論:Dysferlin肌病易誤診和漏診。肌肉組織活檢特異性dysferlin顯著降低或缺如、DYSF基因突變,有助于明確診斷和分型診斷。
【關鍵詞】Miyoshi;dysferlin;dysferlinopathy;遠端肌病;肌營養不良
【中圖分類號】R746 【文獻標識碼】A 【DOI】10.12332/j.issn.2095-6525.2020.12.235
1 臨床資料
患者男性,21歲,以雙下肢進行性無力為首發癥狀,病程4年,表現為平地行走耐力降低,行走拖步改變,足跟、足尖行走不能,癥狀緩慢加重,2年前逐漸出現上樓、蹲起困難,行走跨域步態,無明顯上肢無力,無面部和頸部肌無力,無發聲及吞咽困難,無呼吸困難、胸悶,無肌痛、僵硬、肉跳等;既往無特殊疾病史;患者為第1胎第1產,足月順產,無產傷史和窒息史,無圍生期特殊疾病病史,生長發育里程碑正常;父母非近親婚配,身體健康;家族中無類似疾病病史,無其他遺傳性疾病病史。
體格檢查:神志清楚,高級認知功能正常,語言表達清晰,腦神經檢查未見明顯異常;雙上肢肌力5級,雙下肢近端肌力5-級、遠端4級,雙上肢肌容積、肌張力、腱反射正常,雙側下肢輕度肌萎縮,足內肌萎縮不明顯,未見明顯足下垂,高足弓表現,張力下降,下肢腱反射減弱,雙側指鼻試驗穩準,跟.膝.脛試驗穩準,感覺系統無明顯異常,病理反射未引出。
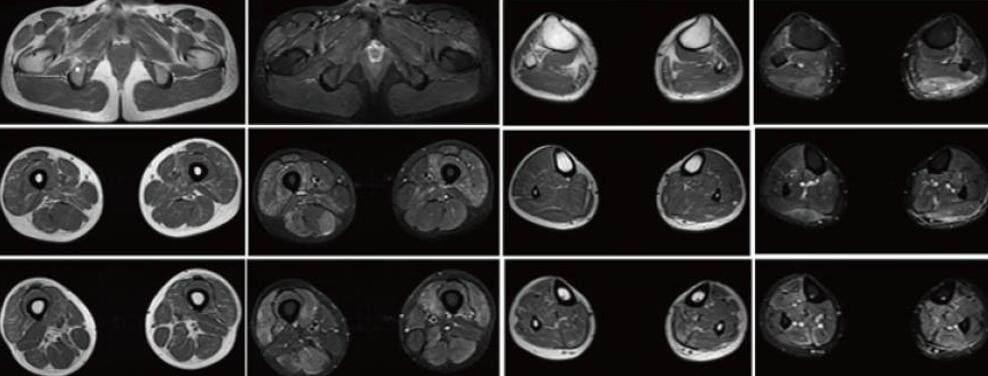
實驗室檢查:生化:ALT 104.10 U/L(9-50),AST 95.60 U/L(15-40);乳酸 3.96mmol/L(0.6-2.2);甲功T3FT4TSH:TSH 4.77mIU/L(0.27-4.20),肌酶全套:CK 6112.0U/L(50-310) CK-MB 160.70U/L(0-25) LDH 470.0U/L(120-250)HBDH 282.0U/L(72-182);ANA抗體譜:抗Ro 52抗體可疑陽性,Sm/RNP可疑陽性;ACA、CCP、ANCA、類風濕因子未見異常;血常規、尿常規、大便常規、凝血功能、乙肝、傳染病三項未見異常。1周后復查肌酶全套:(CK)2949.0U/L(50-310U/L) CK-MB 84.7U/L(0-25) LDH54.0U/L(120-250)HBDH225.0U/L(72-182);神經電生理檢查:NCV:上下肢所檢神經均未見異常。F波:右正中神經、左脛神經潛伏期未見異常,出現率分別為90.0%、100%(正常參考值≥79.0%)。H反射:左脛神經、右正中神經未見異常。EMG:①右股四頭肌安靜時可見1+纖顫電位,小力收縮平均時限縮窄,波幅降低,肌肉輕度收縮時多相電位增多,大力收縮呈病理干擾相等典型肌源性損害改變。? ②左三角肌安靜時未見自發電位,小力收縮平均時限縮窄,波幅降低,大力收縮呈混合相。影像學檢查:磁共振成像顯示下肢肌肉不僅存在萎縮脂肪化,還存在明顯水腫信號;小腿腓腸肌、比目魚肌明顯萎縮,脛骨后肌、拇長屈肌萎縮,且主要以小腿中下部明顯,壓脂像相應肌肉水腫信號,甚至脛前肌也可見水腫信號,大腿股四頭肌、大收肌萎縮,而縫匠肌、股薄肌、半腱肌、股二頭肌長頭相對保留,壓脂像相應肌肉水腫信號。(圖3)。
肌肉病理學檢查:經知情同意,患者自愿行右側肥腸肌肌肉活體組織檢查,結果示肌肉組織形態均呈肌營養不良改變,可見肌纖維大小不一,部分肌纖維萎縮、變性、壞死,結締組織增生,可見炎性細胞浸潤,未見鑲邊空泡,肌纖維糖原和脂肪滴含量正常。免疫組織化學染色顯示,抗肌萎縮蛋白(dystrophin)肌纖維膜均勻、連續,表達.而dysferlin水平明顯缺如(圖2)。
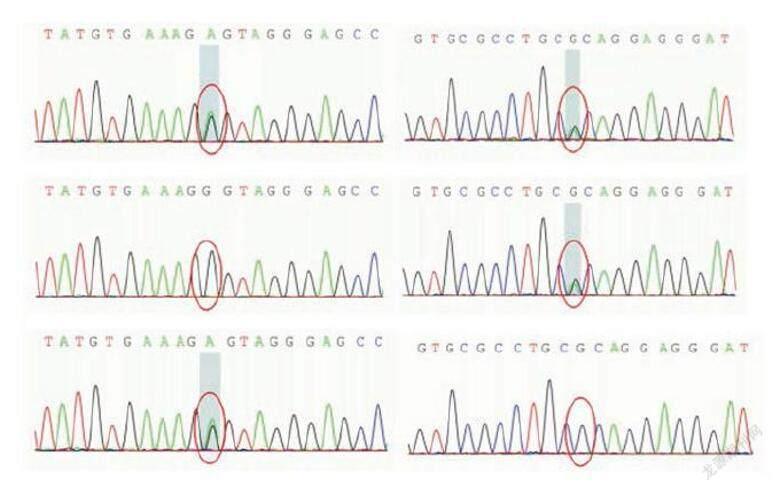
基因檢查:二代測序顯示為dysferlin基因的復合雜合突變,c.3137G>A(鳥嘌呤>腺嘌呤),導致氨基酸改 變p.R1046H(精氨酸>組氨酸)c.5525G>A(鳥嘌呤>腺嘌呤),導致氨基酸改變 p.G1842D(甘氨酸>天冬氨酸),家系驗證顯示分別來自父母雙方,此兩處雜合突變既往均被報道為dysferlin基因的致病突變(圖4)。
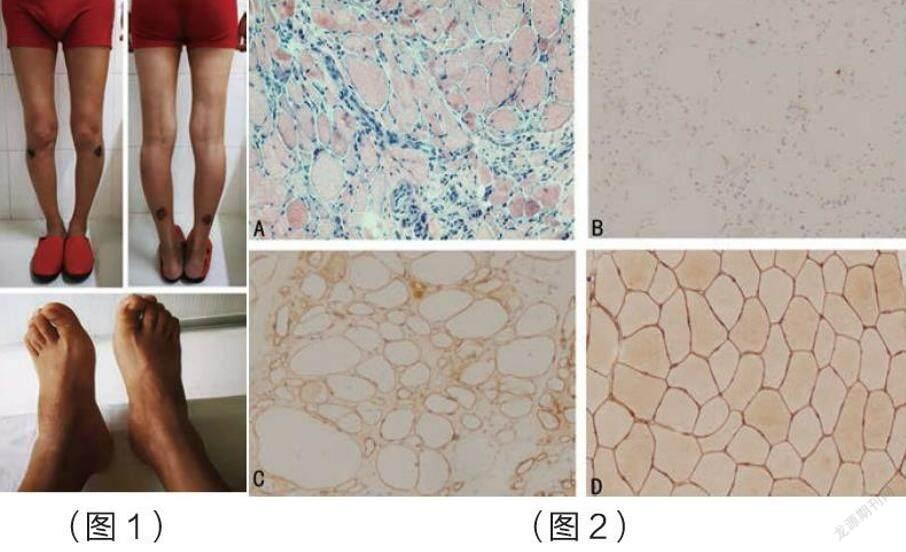
(圖1)患者雙下肢萎縮不僅僅累計雙側小腿,大腿肌肉也有輕度萎縮表現,足內肌萎縮不明顯,未見明顯足下垂,高足弓表現。
(圖2)(A)蘇木精和伊紅染色:(1)肌細胞大小不等,結締組織增生;(2)炎癥細胞侵入壞死肌纖維;(3)肌間小血管炎癥細胞浸潤;
(B)患者dysferlin染色顯示沒有正常的dysferlin染色;
(C)患者dystrophin-R染色顯示正常的dystrophin-R染色
(D)正常人dysferlin染色對照;
(圖3)磁共振成像顯示下肢肌肉不僅存在萎縮脂肪化,還存在明顯水腫信號
(圖4):(上:先癥者)c.3137G>A(鳥嘌呤>腺嘌呤),導致氨基酸改變p.R1046H(精氨酸>組氨酸;c.5525G>A(鳥嘌呤>腺嘌呤),導致氨基酸改變 p.G1842D(甘氨酸>天冬氨酸);(中:父)c.3137G>A(鳥嘌呤>腺嘌呤),導致氨基酸改 變p.R1046H(精氨酸>組氨酸);(下:母)c.5525G>A(鳥嘌呤>腺嘌呤),導致氨基酸改變 p.G1842D(甘氨酸>天冬氨酸);
2 討論
Dysferlin肌病系DYSF基因突變導致的常染色體隱性遺傳性骨骼肌疾病,根據早期受累肌群不同,主要分為2種臨床表型,即以四肢近端肌、盆帶肌受累起源的肢帶型肌營養不良癥2B型(LGMD2B)[1-3]、以腓腸肌受累起源的Miyoshi遠端型肌營養不良癥(MM)[1, 3-5],其他少見臨床表型包括以脛骨前肌受累起源的遠端型肌病(DMAT),近端和遠端同時受累的近遠端型肌病、先天性肌病、腰肌無力型和無癥狀高肌酸激酶血癥[1, 3-5]。不同臨床分型反映同一疾病早期癥狀的差異,同時也造成臨床診斷的復雜化。
MM型以腓腸肌無力發病,表現為足尖行走不能,病情進展相對迅速,發病10年后四肢肌肉廣泛受累,喪失行走能力,晚期累及頸部和面部肌肉[7, 8];而本研究中患者總病程為4年,癥狀表現為下肢無力,行走能力尚存,但從影像學可以看出患者受累已不僅僅局限于下肢遠端,進一步癥狀有待長期隨訪,LGMD2B型多以下肢近端肌無力發病,表現為蹲起困難,逐漸累及上肢近端,病情進展相對緩慢,常于40歲后仍保留行走能力,晚期四肢肌肉廣泛受累,但不累及頭部、面部、頸部肌肉和心肌,不影響認知功能和生存期[2, 8];在本研究中,患者首發癥狀是下肢遠端進行性無力,肌肉萎縮以小腿后部肌群為主,故考慮為MM型,然而患者就診時期肌肉MRI:小腿腓腸肌、比目魚肌萎縮明顯,脛骨后肌、拇長屈肌也同樣萎縮,且主要以小腿中下部明顯,壓脂像相應肌肉水腫信號,甚至脛前肌也可見水腫高信號,大腿股四頭肌、大收肌萎縮明顯,而縫匠肌、股薄肌、半腱肌、股二頭肌長頭相對保留,壓脂像相應肌肉水腫信號。與既往報道受累肌群基本一致[9],但患者雙下肢受累肌群廣泛,由此可見影像學無法區分各種亞型[10],這可能是dysferlinopathy具有統一的病理生理學,因此影像學表現類似,且Dysferlin肌病各亞型進展至晚期,由于廣泛骨骼肌受累,分型診斷較為困難。
除肌營養不良改變外,Dysferlin肌病常表現為不同程度的炎性細胞浸潤,且發病年齡、病程和病變程度均與炎性肌病相似。易誤診為多發性肌炎(PM)[11, 12],有些患者錯誤地長期應用糖皮質激素甚至免疫抑制劑治療。因此,對于激素治療效果欠佳的炎性肌病患者,應行免疫組織化學染色,以鑒別Dysferlin肌病與炎性肌病。
Dysferlin肌病的致病基因是DYSF基因,定位于染色體2p13,包含55個外顯子,DYSF基因突變類型有多種,迄今尚未發現明顯的突變熱點,臨床表型各異,目前尚未發現臨床表型與基因型的關聯性[11, 12]。同一個家系具有相同的遺傳背景,可能有不同的表型[6]。因此,Dysferlin肌病臨床和遺傳具有高度異質性,本研究患者為散發病例,其基因為dysferlin基因的復合雜合突變,分別來自父母雙方,此兩處雜合突變既往均被報道為dysferlin基因的致病突變。結合患者臨床及最先受累肌群考慮為MM型。
關于Miyoshi遠端肌病的治療目前尚無特效療法,主要為對癥治療。 本例患者曾用激素治療1月余,聯合輔酶Q10、艾地苯醌、復合維生素B等治療后癥狀無緩解。
綜上所述,我們報道了一例貴州地區散發型Miyoshi遠端型肌營養不良癥,其臨床特征與既往報道基本一致,但Dysferlin肌病存在臨床異質性,易誤診和漏診。詳細的病史詢問、肌肉組織活檢特異性dysferlin顯著降低或缺如、DYSF基因突變,有助于明確診斷和分型診斷。
參考文獻:
[1]Nguyen, K., et al., Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Human Mutation, 2005. 26(2): p. 165-165.
[2]Mahjneh, I., et al., Dysferlinopathy (LGMD2B): A 23-year follow-up study of 10 patients homozygous for the same frameshifting dysferlin mutations. Neuromuscular Disorders, 2001. 11(1): p. 20-26.
[3]C, et al., Redefining dysferlinopathy phenotypes based on clinical findings and muscle imaging studies. Neurology, 2010.
[4]Cupler, E.J., et al., Miyoshi myopathy in Saudi Arabia: Clinical, electrophysiological, histopathological and radiological features. Neuromuscular Disorders Nmd, 1998. 8(5): p. 321-326.
[5]Cho, H.J., et al., Clinical and genetic analysis of Korean patients with Miyoshi myopathy: identification of three novel mutations in the DYSF gene. J Korean Med Sci, 2006. 21(4): p. 724-7.
[6]張惠麗等, Dysferlin肌病兩家系三例臨床表型及基因突變分析.中國現代神經疾病雜志, 2018. 18(07): 514-519.
[7]A novel compound heterozygous dysferlin mutation inMiyoshi myopathy.
[8]Omar, A.M.X.J., Limb-girdle muscular dystrophy subtypes First-reported cohort from northeastern China. 中國神經再生研究:英文版, 2013. 8(20): p. 1907-1918.
[9]Diaz-Manera, J., et al., Muscle MRI in patients with dysferlinopathy: Pattern recognition and implications for clinical trials. J Neurol Neurosurg Psychiatry, 2018. 89(10).
[10]金蘇芹等, Muscle magnetic resonance imaging changes and relationship with clinical symptoms in patients with dysferlinopathy%dysferlinopathy 患者大腿骨骼肌 MRI 改變及其與臨床表現的相關性.中華神經科雜志, 2014. 000(006): 412-417.
[11]胡靜袁軍輝李娜趙哲沈宏銳, dysferlinopathy患者八例臨床及分子病理學特點. 中華神經科雜志, 2007. 40(12): 807-811.
[12]奚劍英等, Dysferlin肌病的臨床和病理特點分析(附6例報道). 中國臨床神經科學, 2007(03): 296-301.
[13]Bansal, D., et al., Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature, 2003. 423(6936): p. p. 168-172.
