Fabry病的診斷(附1例報告)
2020-10-13 14:23:22石國英徐紅李娟
山東醫(yī)藥 2020年27期
石國英,徐紅,李娟
1陜西省中醫(yī)醫(yī)院,西安710003;2 空軍軍醫(yī)大學西京醫(yī)院;3西安市兒童醫(yī)院
Fabry病為一種罕見的X染色體連鎖隱性遺傳性疾病,發(fā)病率為1∶110 000~1∶40 000。Fabry病致病基因定位在Xq22,致病基因編碼為α-半乳糖苷酶A,由于基因突變導致細胞溶酶體中α-半乳糖苷酶A功能部分或全部缺失,導致三己糖酰基鞘脂醇(Gb3)的正常降解途徑受阻,從而貯積在心、肝、眼、腦、腎及皮膚的神經、血管等多種組織細胞溶酶體中[1],引發(fā)相應器官的功能障礙。Fabry病屬于溶酶體貯積癥的一種,溶酶體貯積癥可導致炎癥的激活,而慢性炎癥的激活會導致纖維化,從而引起多器官損傷[2]。Fabry病臨床表現(xiàn)多樣,癥狀不一,臨床上容易誤診、漏診。現(xiàn)回顧性分析1例Fabry病患者的臨床表現(xiàn)、腎臟病理結果、電鏡結果、骨髓穿刺結果,總結其診斷要點,以提高對本病的認識。
1 資料分析
患者,男,23歲,2014年9月23日因發(fā)現(xiàn)尿中泡沫增多2年,遂來本院就診,患者于1996年發(fā)現(xiàn)前胸漸發(fā)針尖樣紫紅色皮疹,按之不褪色,不伴瘙癢,伴有間斷腹瀉。于西京醫(yī)院行皮膚活檢,病理檢查:表皮角化過度,棘層肥厚,表皮突包繞瘤體,真皮淺層可見擴張血管,管腔內見大量紅細胞。病理結果:血管角質瘤。給予對癥治療(具體用藥不詳)。2012年因腹瀉,尿中泡沫增多就診于烏魯木齊市第四附屬醫(yī)院,行結腸鏡檢查未見異常改變,尿常規(guī)顯示尿蛋白(+++),給予中藥調理半年余(具體用藥不詳),之后一直未復診。多年來皮疹向軀干及四肢擴散(圖1),伴無汗,四肢燒灼樣疼痛,尿中泡沫增多,仍間斷伴有腹瀉。患者母親有少汗及四肢疼痛發(fā)作史。入院檢查:精神可,表情自如,四肢及軀干散在紫紅色針尖樣丘疹。實驗室檢查:尿常規(guī)顯示尿蛋白(+++),血清肌酐195 μmol/L。結合患者病史及家族史,考慮Fabry病。為求明確診斷行腎活檢穿刺術。所取腎組織分別行光鏡、免疫熒光及電鏡檢查,并行骨髓穿刺觀察骨髓象。光鏡結果:光鏡下可見2條腎皮質、13個腎小球、4個缺血性球性硬化、8個節(jié)段性硬化伴球囊粘連,另見腎小球系膜細胞和基質輕度增生,上皮細胞增生、肥大;腎小管上皮細胞空泡及顆粒變性,片狀萎縮;腎間質片狀纖維化伴淋巴及單核細胞浸潤;小動脈管壁增厚。診斷結果:局灶節(jié)段性腎小球硬化癥(NOS型)(圖2)。免疫熒光結果:陰性。電鏡結果:電鏡下僅見1個硬化腎小球,殘留細胞胞質內查見髓樣小體,腎小管萎縮,腎間質纖維化,腎間質細胞胞質內亦可見髓樣小體(圖3)。骨髓穿刺結果:瑞氏—姬姆薩染色,油鏡下觀察:骨髓增生活躍,粒系占50.50%,紅系占37.00%,成熟淋巴細胞占9.50%,漿細胞占1.00%,單核細胞占0.50%,可見貯脂細胞,占1.5%;細胞大,圓形或不規(guī)則多邊形,細胞核小,偏于細胞一側,染色質呈網(wǎng)狀,胞質豐富,淡藍色,胞質內充滿大小不一,透明的脂質顆粒,呈泡沫狀或蜂窩狀(圖4)。結合光鏡、免疫熒光、電鏡、骨髓穿刺結果,診斷為Fabry病。患者入院期間西醫(yī)治療予保腎、降蛋白、降壓等對癥治療,中醫(yī)以健脾益腎、活血化瘀為主,經治療后病情好轉出院,欲擇期行腎移植手術。
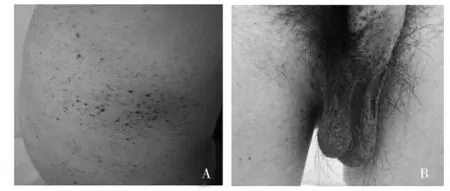
注:A:軀干部針尖至粟粒大暗紫紅色丘疹; B:陰囊部針尖至粟粒大暗紫紅色丘疹。
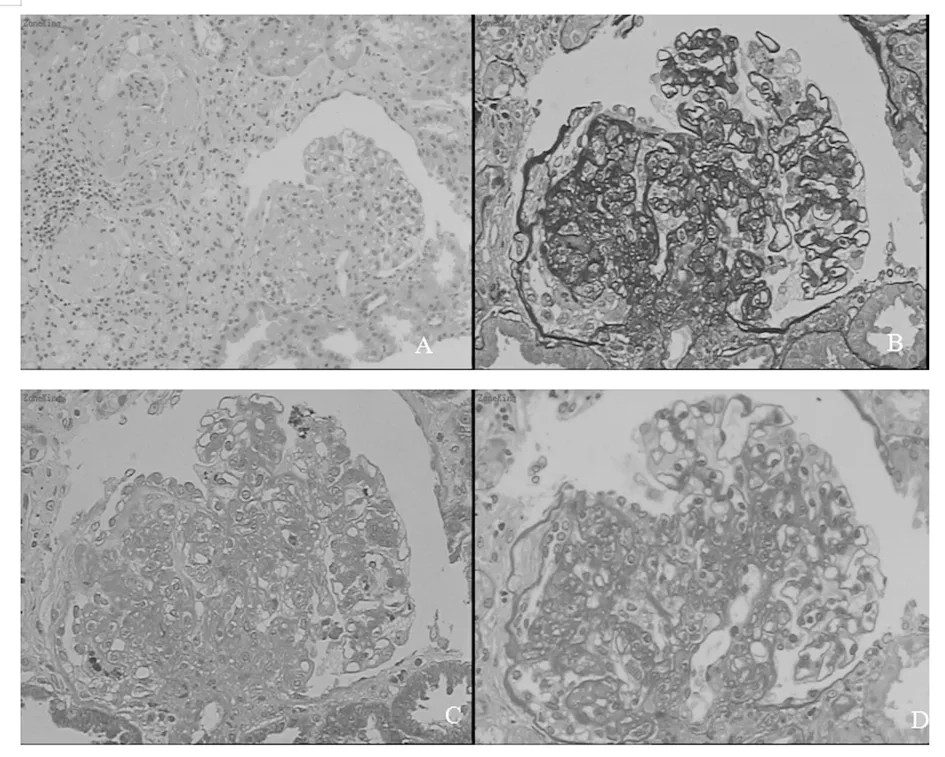
注:A:HE染色(×20);B:六胺銀染色(×40);C:masson 染色(×40);D:PAS染色(×40);腎小球呈FSGS改變。

注:A:TEM(×5 000),B:TEM(×10 000);胞質內見髓樣小體。
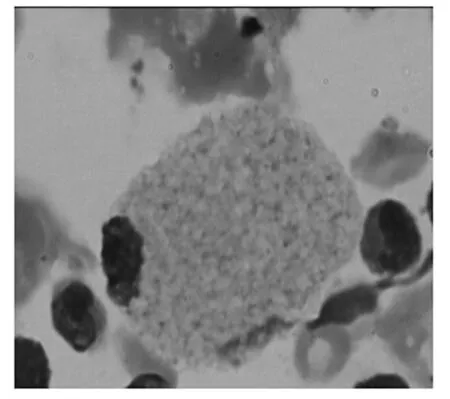
注:貯脂細胞:細胞大,細胞核小,胞質豐富,胞質內充滿大小不一、透明的脂質顆粒、呈泡沫狀或蜂窩狀。
2 討論
Fabry病是1898年由英格蘭的Andeson和德國的Fabry同時報道,又稱安德森—法布里病。此病是一種罕見的X染色體連鎖隱性遺傳性疾病,病變可累及全身多個器官和系統(tǒng),臨床早期多以皮膚受損為主要癥狀,后期逐漸累及心、肝、眼、腦及腎臟,表現(xiàn)為彌漫性軀體血管角質瘤、皮膚無汗或少汗、發(fā)熱、角膜及晶狀體渾濁、間歇性肢端疼痛、體力下降、輕度蛋白尿和胃腸道癥狀[3,4]。
Fabry病的血管角質瘤分5型,即肢端型、陰囊型、丘疹型、界限型和軀體彌漫型,軀體彌漫型血管角質瘤為少見的先天性溶酶體貯積病的皮膚標志,本病十分罕見,大部分患者早期表現(xiàn)為皮膚損害,多就診于皮膚科,常被誤診,因此臨床遇到血管角質瘤的患者,尤其是軀體彌漫型血管角質瘤,應追問家族史,對患者進行進一步檢查,并定期隨訪。若患者出現(xiàn)蛋白尿、腎功能異常,要引起足夠的重視,除外先天性溶酶體貯積病,如Fabry病。Fabry病后期累及腎臟,腎穿刺活檢光鏡檢查發(fā)病早期表現(xiàn)為系膜增生病變,以后逐步進展為局灶節(jié)段腎小球硬化樣病變,甚至腎小球硬化,這個過程是不可逆的,最終引起終末期腎功能衰竭,從而導致死亡,這是Fabry病死亡的最常見原因。因此有研究指出在終末期腎臟疾病患者中,早期診斷和治療在腎臟疾病管理中至關重要[5]。本研究中,患者最早以皮膚紫紅色皮疹發(fā)病,病理診斷為血管角質瘤,后因出現(xiàn)蛋白尿,腎功能異常,入住本院腎內科。追問其家族史,發(fā)現(xiàn)其母親有少汗及四肢疼痛發(fā)作史,行全身檢查,經腎穿刺活檢及骨髓穿刺檢查,診斷為Fabry病。
Fabry病的診斷主要依靠經典的臨床表現(xiàn)及相應的組織病理檢查。臨床上Fabry病除特征性的皮膚及神經系統(tǒng)受累表現(xiàn)外,其他器官受累均缺乏特征性表現(xiàn),患者發(fā)病年齡及表現(xiàn)方式不同,病變后期多累及腎臟,需要行腎穿刺活檢,但腎穿刺檢查光鏡下改變常無特異性診斷價值,電鏡下相應的腎組織細胞質內查見髓樣小體,該小體呈較高電子密度,圓形或卵圓形,小體內部呈層狀,形似斑馬皮,亦似洋蔥皮或髓鞘結構,故稱髓樣小體,為Fabry病特征性表現(xiàn)[6],是診斷該疾病的重要依據(jù)。骨髓涂片中發(fā)現(xiàn)貯脂細胞可進一步明確疾病診斷。因此,單純依靠臨床表現(xiàn)及光鏡檢查,不能準確診斷Fabry病,極易漏診誤診,同時行腎組織活檢電鏡檢查及骨髓穿刺檢查,是減少漏診和誤診的重要方法。
Fabry病需要與局灶節(jié)段腎小球硬化癥(FSGS)、慶大霉素的腎毒性、尼曼-匹克病等相鑒別。FSGS是一組以大量蛋白尿和腎病綜合征為主要臨床特征,病理以局灶和節(jié)段分布的硬化性病變?yōu)橹饕兓哪I小球疾病。免疫熒光檢查常為陰性,光鏡下腎小球局灶節(jié)段性硬化,足細胞空泡變性,故Fabry病最易誤診為FSGS,但電鏡下FSGS足細胞內無髓樣小體。劉東娟等[7]通過建立慶大霉素腎毒性模型,應用透射電鏡技術對腎臟近端小管上皮細胞的凋亡進行超微結構的觀察,在用藥10 d后發(fā)現(xiàn)大鼠近端腎小管上皮細胞質溶解,胞質內線粒體腫脹,嵴減少或消失呈空泡樣,并向核周圍聚集,粗面內質網(wǎng)擴張,溶酶體增多,并見髓樣小體形成,此特點與Fabry病的電鏡下表現(xiàn)相似,需要詳細詢問患者臨床用藥史并結合臨床表現(xiàn),必要時行腎穿刺活檢才能排除。尼曼—匹克病是由于鞘磷脂酶活性降低,引起的鞘磷脂質貯積病,以嬰幼兒及兒童起病多見,神經鞘磷脂和膽固醇在組織中過度沉積,主要受累部位為肝、脾及骨髓[8]。骨髓穿刺可查見泡沫細胞,與Fabry病難區(qū)分,但尼曼—匹克病常見肝脾大,無皮膚癥狀及腎臟受累,電鏡檢查無髓樣小體。
由于Fabry病晚期多累及腎臟,引起腎功能衰竭。腎移植可以緩解Fabry病的終末期腎臟的臨床癥狀,但目前國內器官來源短缺,報道較少。有研究[9]表明,男性患者、大量蛋白尿、伴有小管間質病變或多系統(tǒng)損害者,腎移植并不能提高患者生存率。現(xiàn)在多提倡特異性治療,即酶替代療法。研究發(fā)現(xiàn),大部分Fabry病患者的α-半乳糖苷酶A活性缺乏,由于蛋白質的錯誤折疊,酶無法輸送到溶酶體,突變蛋白滯留在內質網(wǎng)或過早的降解所致[10]。用重組α-半乳糖苷酶A進行酶替代治療,可有效改善Fabry病患者癥狀,減少Gb3在血管內皮細胞的貯積,改善心、腦、腎血管的功能,提高生活質量,預防晚期并發(fā)癥的發(fā)生[11]。
總之,F(xiàn)abry病臨床上男性多于女性,表現(xiàn)為多器官受累,腎臟是最常受累器官,腎衰竭是其主要死亡原因,腎穿刺組織電鏡檢查見髓樣小體是該疾病的確診依據(jù),家族遺傳史及骨髓涂片中發(fā)現(xiàn)貯脂細胞可作為診斷的參考。Fabry病的臨床表現(xiàn)多種多樣,且病死率高,需引起各科醫(yī)生重視,熟悉該病臨床特征,提高診治水平。
