鞍區朗格漢斯細胞組織細胞增生癥臨床特點分析
2020-08-07 02:49:12王志成朱建宇張毅李曉旭劉杰朱惠娟姚勇潘慧鄧侃
中國現代神經疾病雜志 2020年7期
王志成 朱建宇 張毅 李曉旭 劉杰 朱惠娟 姚勇 潘慧 鄧侃
朗格漢斯細胞組織細胞增生癥(LCH)是一種病因不明的組織細胞浸潤性疾病,因腫瘤細胞在組織學形態和免疫表型與皮膚和黏膜朗格漢斯細胞相似而得名,但是從基因表達陣列數據看,病態朗格漢斯細胞起源于髓樣樹突狀細胞,并非由皮膚和黏膜朗格漢斯細胞轉化而來[1]。LCH好發于兒童和青少年,可以累及單器官或多器官,依次為骨骼(80%)、皮膚(33%)、垂體(25%)、肝臟(15%)、脾(15%)、造血系統(15%)、肺(15%)、淋巴結(5%~10%)和中樞神經系統除垂體外的其他部位(2%~4%)[2]。受累部位不同,臨床癥狀略有差異,可以表現為發熱、乏力、消瘦、骨痛、皮損、中樞性尿崩癥、黃疸、咳嗽、胸痛等,明確診斷依靠組織病理學檢查。累及鞍區的LCH 臨床罕見,主要表現為中樞性尿崩癥、頭痛、視力視野損害、腺垂體功能減退癥、下丘腦相關癥狀等,其中中樞性尿崩癥為最常見癥狀。對于以中樞性尿崩癥發病、頭部MRI 提示鞍區占位性病變、擬診LCH 的患者,考慮到腦組織活檢術的風險相對較高,積極尋找可能支持診斷的顱外病變即顯得十分重要。單純累及鞍區者十分罕見,在以中樞性尿崩癥發病的LCH 中僅占9%[3],并且既往多為個案報道。加之缺少特異性臨床表現和可用于組織活檢的顱外病變,大多數患者僅能通過定期隨訪,直至鞍區病變進展而不得不接受組織活檢、或出現顱外病變、或接受經驗性治療并經MRI監測病情變化。本文擬對中國醫學科學院北京協和醫院神經外科近年收治的8例累及鞍區的LCH病例的臨床特點進行總結,并探討鞍區組織活檢術的安全性。
臨床資料
通過中國醫學科學院北京協和醫院信息系統檢索獲得2011 年11 月至2019 年11 月在神經外科住院并符合以下納入標準的病例共計8 例:頭部MRI 提示鞍區占位性病變;于神經內鏡下行經鼻蝶入路(ETA)或擴大經鼻蝶入路(EETA)鞍區病變組織活檢術證實為LCH。男性4 例,女性4 例;年齡為10 ~ 47 歲,中位年齡15.50(12.25,38.00)歲,其中 <18 歲者 5 例、≥ 18 歲 3 例;發病至確診時間 2 ~ 50 個月,中位時間4.00(2.25,28.75)個月(表1)。(1)癥狀與體征:主要以多飲、多尿、煩渴、夜尿增多等中樞性尿崩癥表現發病;其中,7 例同時伴有食欲減退、乏力、性欲減退、月經紊亂、閉經、遺精減少等腺垂體功能減退癥狀,1 例伴頭痛,1 例表現為下丘腦相關癥狀如間斷性發熱(37 ~38 ℃),多出現在早餐或午餐后,約至黃昏時(18∶00)自行退熱(表1)。(2)實驗室檢查:所有患者血清和腦脊液甲胎蛋白(AFP)或β-人絨毛膜促性腺激素(β-hCG)水平均于正常參考值范圍(表2)。(3)垂體MRI 表現:占位性病變累及范圍包括鞍內及垂體柄(例2、例6、例8),垂體柄及下丘腦(例5、例7),鞍內、垂體柄及下丘腦(例3),鞍內、左側顳葉內側和胼胝體(例4),以及單純垂體柄受累(例1)。T1WI 神經垂體高信號消失,呈等信號者8 例;T2WI 呈等信號者6 例(例1 ~ 例4、例6、例8)、低信號1例(例5)或混雜信號1例(例7);增強掃描呈均勻強化者6 例(例1、例3 ~例7),不均勻強化者2 例(例2、例8)。8 例患者均未發現顱外病變(表2)。(4)組織活檢術:對于MRI 顯示占位性病變累及鞍內的患者(例2 ~例4、例6、例8)于神經內鏡下經鼻蝶入路行鞍內病變組織活檢術,術前30 min 和術后72 h 常規應用頭孢呋辛鈉1.50 g/次(2 次/d)靜脈滴注預防感染;病變完全位于鞍上者(例1、例5、例7)經神經內鏡下擴大經鼻蝶入路行鞍上病變組織活檢術和脂肪浴缸塞聯合鼻中隔黏膜瓣鞍底重建術,術前30 min 和術后72 h 常規予頭孢他啶2 g/次(2 次/d)靜脈滴注預防感染。(5)術后并發癥:本組僅1例(例5)患者組織活檢術后1 d出現雙眼顳側視物成雙、模糊,術后3 d 上述癥狀緩解,術后6 d 上述癥狀完全消失,考慮為短暫性下丘腦反應。無一例患者術后1 個月內發生腦脊液鼻漏、中樞神經系統感染、非計劃二次手術、死亡等其他手術相關并發癥(表2)。
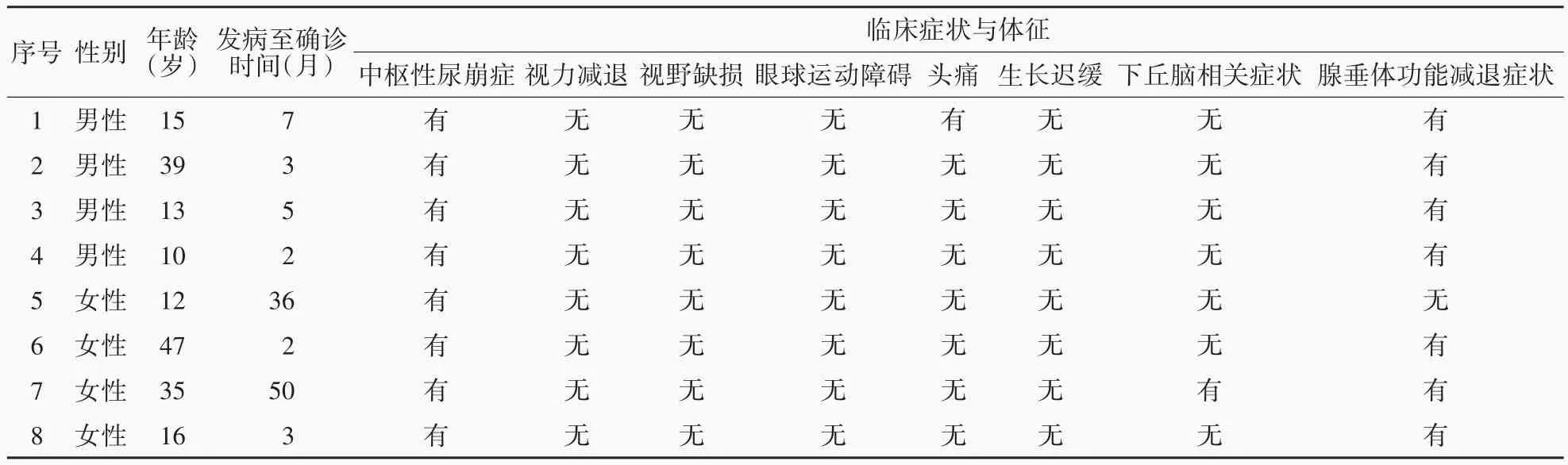
表1 8例鞍區朗格漢斯細胞組織細胞增生癥患者社會人口學資料和臨床表現Table 1. Demographic data and clinical manifestations of 8 patients with sellar LCH
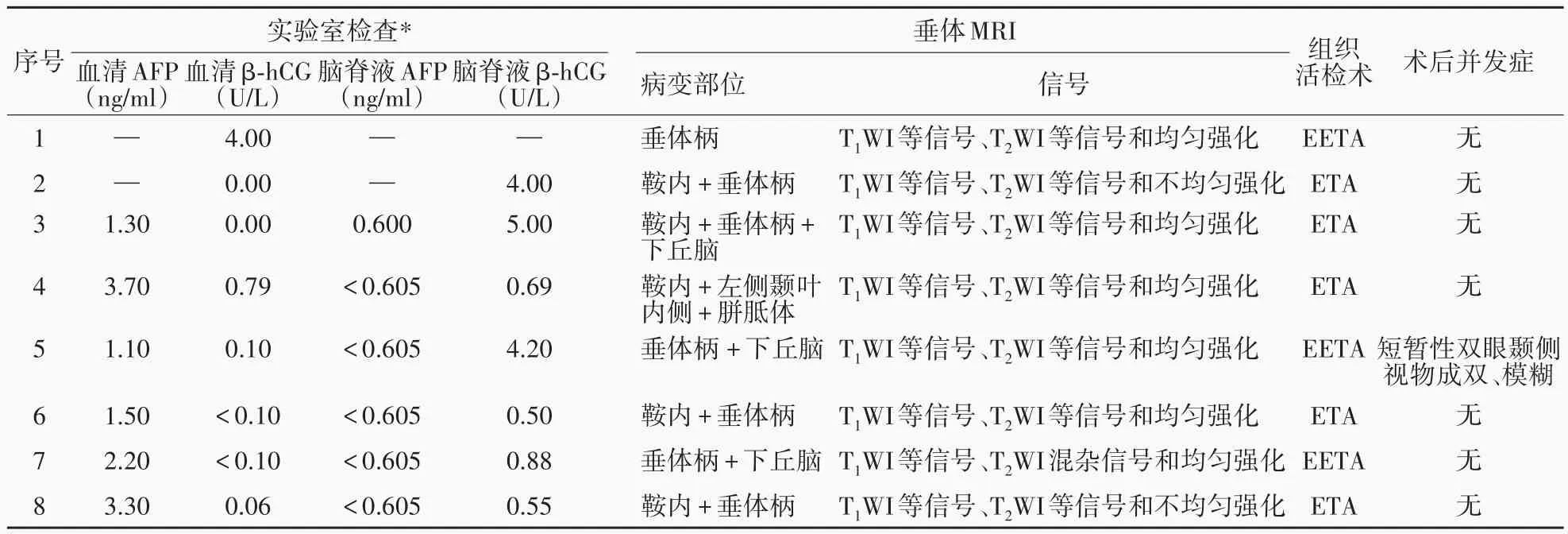
表2 8例鞍區朗格漢斯細胞組織細胞增生癥患者實驗室、MRI、組織活檢術和術后并發癥Table 2. Laboratory, MRI, biopsy and postoperative complications of 8 patients with sellar LCH
典型病例
患者(例8) 女性,16 歲。因多飲、多尿4 個月,月經不規律 2 個月,于 2019 年 4 月 3 日入院。患者入院前4 個月(2018 年12 月)無明顯誘因出現多飲、多尿,每日飲水量約10 L,白天小便1 次/h、夜尿3 ~4次;入院前2 個月開始出現月經不規律,偶有惡心,但無頭痛、嘔吐、發熱、乏力,以及體毛脫落、性欲改變和異常泌乳等改變,自覺無視力下降、視野缺損等癥狀,外院垂體MRI 檢查顯示垂體形態飽滿,信號均勻,冠狀位最高徑約7.50 mm,垂體柄欠清晰,略增粗,視交叉無明顯受壓,為求進一步治療至我院就診。入院后體格檢查未見明顯異常。垂體MRI 顯示垂體飽滿,上緣略膨隆,橫徑12.70 mm、上下徑7.40 mm、前后徑8.90 mm,信號均勻,呈等T1、等T2信號,增強后病灶呈明顯不均勻強化;垂體柄增粗,橫徑5 mm、前后徑5 mm,視交叉未見受壓、移位,神經垂體信號消失,松果體區未見異常信號(圖1)。臨床診斷為垂體柄占位性病變,原因待查。于2019年4月8日經神經內鏡下經鼻蝶入路行鞍內病變組織活檢術,術中于鞍內神經垂體部位可見灰白色病變組織,質地較韌,邊界欠清晰,血運不豐富,向鞍上生長。組織標本鏡下觀察,垂體局部纖維組織增生,可見組織細胞、淋巴細胞及嗜酸性粒細胞浸潤(圖2a);免疫組化染色,上皮細胞表達廣譜細胞角蛋白(AE1/AE3);腫瘤細胞表達CD163 和CD1a(圖2b),局灶性表達Langerin 和S-100 蛋白(S-100),部分表達CD20 和CD3,散在表達CD68,不表達人婆羅雙樹樣基因-4 蛋白(SALL4),Ki-67 抗原標記指數約為10%;最終病理診斷為LCH。患者術后3 天出院,無手術相關并發癥,出院后在外院接受藥物化療(具體方案不詳),術后3 個月隨訪時仍在接受藥物化療。
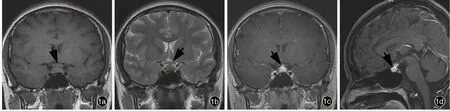
圖1 例8 患者術前垂體MRI 檢查所見 1a 冠狀位T1WI 顯示垂體飽滿,上緣略膨隆,橫徑12.70 mm、上下徑7.40 mm,呈等信號(箭頭所示);垂體柄增粗,橫徑5 mm;視交叉未見受壓移位 1b 冠狀位T2WI 掃描垂體呈等信號(箭頭所示) 1c 冠狀位增強T1WI 掃描,垂體呈不均勻強化、垂體柄明顯強化(箭頭所示) 1d 矢狀位增強T1WI 顯示垂體前后徑8.90 mm、垂體柄前后徑5 mm(箭頭所示),神經垂體信號消失,松果體區未見異常信號Figure 1 Head MRI findings before operation of Case 8 Coronal T1WI showed the pituitary gland was plump, the upper edge was slightly bulged, the transverse diameter was 12.70 mm, and the height was 7.40 mm, with isointense signal (arrow indicates). The pituitary stalk was thickened, the transverse diameter was 5 mm, and the optic chiasm was not compressed (Panel 1a). Coronal T2WI showed the pituitary with isointense signal (arrow indicates, Panel 1b). Coronal enhanced T1WI showed that the enhancement of the pituitary was not even, and the pituitary stalk was obviously enhanced (arrow indicates, Panel 1c). Sagittal enhanced T1WI showed the anterior-posterior diameter of pituitary was 8.90 mm, the anterior-posterior diameter of pituitary stalk was 5 mm (arrow indicates), the high signal at the posterior lobe of pituitary was not shown, and the abnormal signal at pineal region was not found (Panel 1d).
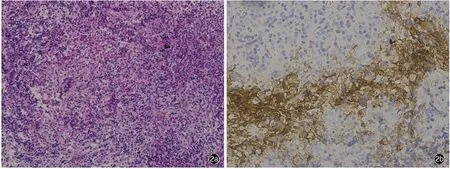
圖2 例8 患者光學顯微鏡觀察所見 2a 垂體局部纖維組織增生,可見組織細胞、淋巴細胞及嗜酸性粒細胞浸潤 HE 染色 ×100 2b 腫瘤細胞表達CD1a 免疫組化染色(EnVison二步法) ×200Figure 2 Optic microscopy findings of Case 8 Local fibrous tissue hyperplasia and infiltration of tissue cells were seen, and lymphocytes and eosinophils were in pituitary (Panel 2a). HE staining × 100 CD1a was positive in tumor cells (Panel 2b).Immunohistochemical staining (EnVison) ×200
討 論
朗格漢斯細胞組織細胞增生癥在兒童和青少年(< 15 歲)人群中的發病率約為4.6/100 萬人年[4],成人發病率為 1 ~ 2/100 萬人年[5],老年人群偶見個案報道[6],考慮到部分局灶性LCH 具有自發緩解特點,其真實年發病率可能高于文獻報道的數據[7]。兒童和青少年(<15 歲)LCH 的確診中位年齡約為3.5 歲,高峰發病年齡 < 1 歲[4]。本組 8 例鞍區 LCH患者的中位年齡為15.5 歲,其中最小患兒為10 歲,可能與LCH 在年齡更小的患者中發生皮損的概率更高有關[8]。
根據文獻報道,約有6%的LCH 患者確診時中樞神經系統已受累[9],尤以中樞性尿崩癥最為常見,而約有15%單純表現為中樞性尿崩癥的患者最終被確診為 LCH[10]。Prosch 等[3]認為,約有 10.23%(127/1242)的LCH 患者存在中樞性尿崩癥,其中42.52%(54/127)確診時即已存在中樞性尿崩癥;在此部分患者中90.74%(49/54)是經顱外病變組織活檢術被確診,其中18.37%(9/49)確診時即已存在顱外病變。本組8 例患者均以多飲、多尿等中樞性尿崩癥發病,男女比例為1∶1,確診前均未發現其他顱外病變,且無一例存在視力下降、視野缺損或眼球運動障礙等癥狀與體征,但有7 例表現有食欲減退、乏力、性欲減退、月經紊亂、閉經、遺精減少等腺垂體功能減退癥狀;病變累及下丘腦區域時,患者可出現嗜食、體溫調節紊亂、嗜睡和短期記憶障礙等下丘腦相關癥狀[11],但本組病變累及下丘腦的3例患者中僅1例出現間斷性發熱性下丘腦反應。
對于存在中樞性尿崩癥的患者,頭部MRI 是首選影像學檢查方法,T1WI 顯示的神經垂體高信號消失被認為是LCH 鞍區受累的典型表現,主要與神經垂體含抗利尿激素的分泌顆粒耗竭有關,本組患者均未見明顯的神經垂體高信號征象。然而,神經垂體高信號消失并非LCH 的特異性表現[12]。從病變部位看,本組垂體柄受累者7 例、鞍內受累者5 例,其中鞍內和垂體柄同時受累(3 例)較為常見,其次為垂體柄和下丘腦(2 例)受累;從病變信號看,本組共有4 例(例1、例 3、例4、例 6)呈現T1WI 等信號、T2WI等信號和均勻強化,1 例(例5)呈T1WI等信號、T2WI低信號和均勻強化,1 例(例7)呈T1WI等信號、T2WI 混雜信號和均勻強化,2 例(例 2、例 8)呈現T1WI 等信號、T2WI 等信號和不均勻強化。遺憾的是,盡管MRI 在鞍區占位性病變的臨床診斷中具有不可替代的作用,可以反映病變部位、累及范圍,并監測疾病進展,但其表現并不具有特異性[13-14]。治療方面,需根據病變累及范圍和病情嚴重程度選擇治療方法,主要包括局部放射治療、全身化療和糖皮質激素治療[11,15]。
朗格漢斯細胞組織細胞增生癥應注意與垂體腺瘤、生殖細胞腫瘤、淋巴細胞性垂體炎等鞍區占位性病變相鑒別。(1)垂體腺瘤:為臨床十分常見的顱內腫瘤,約占顱內腫瘤的15%[16],發病率約為0.1%[17]。垂體腺瘤可由腺垂體中任意一種細胞發展而來,分為功能性垂體腺瘤和無功能性垂體腺瘤,功能性垂體腺瘤主要包括垂體生長激素腺瘤、泌乳素腺瘤、促腎上腺皮質激素腺瘤、促甲狀腺激素腺瘤、促性腺激素腺瘤等[18],進而導致巨人癥和(或)肢端肥大癥、停經和(或)泌乳、Cushing病、繼發性甲狀腺功能亢進癥等,從而表現出相應臨床癥狀。實驗室檢查相關激素水平升高,有助于鑒別診斷。據腫瘤大小可分為垂體微腺瘤(直徑<10 mm)和大腺瘤(直徑≥10 mm)。無功能性垂體腺瘤由于缺乏特異性激素相關癥狀,多以局部占位效應而就診,確診時大多已進展為大腺瘤,主要表現為頭痛、視力下降、視野缺損,以及因壓迫導致的腺垂體功能減退癥狀。由于腫瘤生長較為緩慢,較少導致中樞性尿崩癥。垂體腺瘤T1WI呈等或低信號,T2WI呈等信號,強化程度低于正常垂體[19]。治療方法以外科手術切除或藥物治療為主。(2)顱內生殖細胞腫瘤:較為少見,在中樞神經系統腫瘤中所占比例不足5%、兒童腦腫瘤中占3%~11%,發病率約為0.1/10 萬[20-21],男女比例為4 ~ 5∶1,發病高峰年齡為 10~ 20 歲[22],與 LCH 相似。顱內生殖細胞腫瘤好發于松果體和鞍上區[23],位于松果體區的生殖細胞腫瘤通常表現為堵塞腦脊液通道導致的顱內高壓癥狀,位于鞍上區的生殖細胞腫瘤則主要表現為中樞性尿崩癥、頭痛、腺垂體功能減退癥、生長遲緩、視力障礙和視野缺損等。根據組織學形態分為生殖細胞瘤和非生殖細胞瘤性生殖細胞腫瘤(NGGCTs),后者血清和(或)腦脊液AFP 或β-hCG 水平升高,主要累及松果體區[24]。T1WI 呈等或低信號,T2WI 呈等或高信號,增強后病灶呈明顯強化征象。由于部分生殖細胞腫瘤臨床表現缺乏特異性,其診斷可被延誤數月甚至數年[25-26],組織活檢術為診斷“金標準”[27]。治療方法主要包括放射治療、藥物化療和手術治療[28-30]。(3)淋巴細胞性垂體炎:是臨床較為罕見的自身免疫性疾病,以大量淋巴細胞彌漫性浸潤和纖維化為組織學特征[31];發病率約為0.1/100萬,好發于女性,特別是孕后期和產后早期女性,中位發病年齡為37 歲[32]。臨床表現缺乏特異性,主要表現為中樞性尿崩癥、腺垂體功能減退癥狀、頭痛等,尤以中樞性尿崩癥常見,發生率約為72%[33]。約有96%患者MRI 表現為垂體柄增粗、78%患者可鞍內和鞍上同時受累。臨床診斷較為困難,難以與單純累及鞍區的LCH 和生殖細胞腫瘤相鑒別,明確診斷依靠組織活檢術。治療方法以糖皮質激素和免疫抑制治療為主。
鞍區病變的治療方案可截然不同,因此明確診斷尤為重要。組織活檢術仍是診斷LCH 的“金標準”。考慮到顱內組織活檢術的相對高風險,積極尋找顱外病變十分重要,但若缺乏支持LCH 診斷的顱外病變時,是否應積極進行顱內組織活檢術,以及何時進行活檢術即成為診斷與治療的關鍵。Prosch 等[34]認為,盡管同時存在諸多看似典型的臨床表現,也不應在進行組織病理學檢查之前進行臨床診斷并治療。本組8例患者采用神經內鏡下經鼻蝶入路或擴大經鼻蝶入路鞍區病變組織活檢術,術后僅1 例出現短暫性下丘腦反應,無一例并發腦脊液鼻漏、中樞神經系統感染、非計劃二次手術、死亡等不良事件。與開顱手術相比,神經內鏡下經鼻蝶入路組織活檢術具有美觀、對周圍腦組織牽拉小、視野開闊和手術損傷小等優勢[35-36],因此神經內鏡下經鼻蝶入路鞍區占位性病變組織活檢術是一種相對安全的方法。我們的臨床經驗是:(1)對于存在中樞性尿崩癥或MRI 提示鞍區占位性病變,擬診LCH 的患者,應首先完善臨床病史、體格檢查、血清和腦脊液腫瘤標志物、全身骨顯像、頭部和胸部X線、腹部超聲等檢查,積極尋找可能支持LCH 診斷的顱外病變。(2)當缺少顱外病變且鞍區病變直徑<5 mm、無視神經壓迫癥狀時,應密切隨訪,定期復查MRI,一旦內分泌或影像學檢查提示病情進展,應盡早行鞍區病變組織活檢術。(3)對于缺少顱外病變且鞍區病變直徑≥5 mm 或存在視神經壓迫癥狀的患者,除非活檢術風險極高,否則均應盡早施行病變組織活檢術以明確診斷。
鞍區病變病因多樣,部分疾病缺乏特異性臨床表現,難以通過無創性檢查與單純累及鞍區的LCH相鑒別,組織活檢術仍是診斷的“金標準”。神經內鏡下經鼻蝶入路和擴大經鼻蝶入路鞍區病變組織活檢術相對安全、有效,可以為臨床制定最佳治療方案和判斷預后提供依據。
利益沖突無
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
中國生殖健康(2019年3期)2019-02-01 06:12:26
獸醫導刊(2016年6期)2016-05-17 03:50:35
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
