七-(2,6-二甲基)-β -環糊精對喜樹堿增溶作用的機理
2020-06-04 09:51:32韓東旭李昊原辛士剛張洪波
沈陽師范大學學報(自然科學版) 2020年2期
關鍵詞:結構
于 湛, 韓東旭, 李昊原, 辛士剛, 張洪波
(1. 沈陽師范大學 化學化工學院, 沈陽 110034; 2. 沈陽師范大學 實驗教學中心, 沈陽 110034)
0 引 言
環糊精(cyclodextrin,CD)是以包含6個(α-)、7個(β-)或8個(γ-)吡喃葡萄糖為基本結構單元,并由α-1,4糖苷鍵鏈接而成的環狀寡糖[1]。環糊精具有“內腔疏水,外部親水”的特性,外形呈現上窄下寬、兩端開口的中空結構特點。正是由于具有上述結構特點,CD可作為主體與多種無機或有機小分子化合物形成復合物[2]。天然CD特別是β-CD由于具有較強的分子內氫鍵作用,因此在水中溶解度很小。CD的2-、3-或6-羥基具有一定化學活性,可在一定條件下被其他官能團所取代,生成諸如七-(2,6-二甲基)-β-環糊精(DM-β-CD)、七-(2-羥丙基)-β-環糊精(2HP-β-CD)與6-葡萄糖基-β-環糊精(6Glu-β-CD)等CD衍生物。這些CD衍生物在保留了空腔疏水性的前提下,極大地提高了溶解度與選擇性,因此具有良好的應用前景[3-6]。
喜樹堿(camptothecin,CPT)是一種由珙桐科植物喜樹的果實或根莖表皮中提取的細胞毒性喹啉類生物堿。CPT是被用于早期中醫治療癌癥的一種藥物,由于其對DNA拓撲異構酶I具有抑制作用[7],成為早期唯一的一種能夠作用于DNA拓撲異構酶I的抗腫瘤藥物。由于具有抗腫瘤的功效,CPT廣泛應用于直腸癌、腸癌、胃癌和白血病等疾病的治療[8],同時也有一定的副作用,例如嘔吐、腹瀉[9]及血性膀胱炎等[10]。盡管CPT具有很好的藥用前景,但是由于其結構中親水性極性基團數量較少,在水中的溶解度較低,口服后在人體內的吸收利用率不高。
CD可通過與難溶性小分子藥物形成復合物的方式有效提高藥物的溶解度[11]。為了提高CPT水溶性,使其能更好地被生物體吸收,本文使用α-CD、β-CD、γ-CD、2HP-β-CD、DM-β-CD與6Glu-β-CD等6種CD作為增溶劑,研究它們對CPT的增溶作用,并通過分子模擬計算推測了增溶機理。
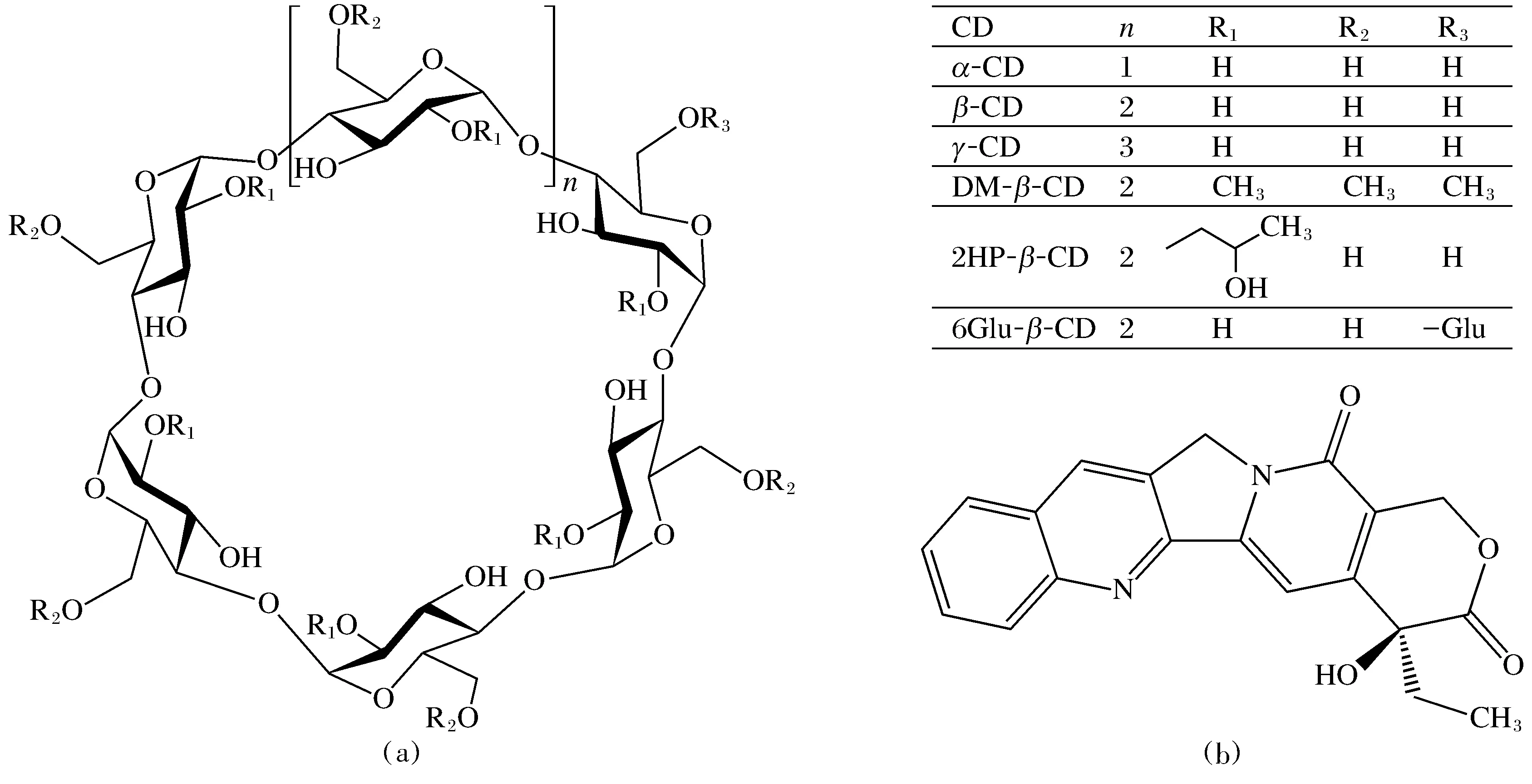
圖1 文中所用CD(A)與CPT(B)的化學結構Fig.1 Chemical structures of CDs (A) and CPT (B) used in this work
1 實驗部分
1.1 儀器與試劑
傅里葉變換紅外光譜儀(NICOLET 380,美國賽默飛),紫外-可見光譜儀(UH5300,日本日立),高速離心機(CT6TA,中國天美)。
CPT購買于西安開來生物制藥有限公司,α-CD、β-CD、γ-CD、DM-β-CD、2HP-β-CD和6Glu-β-CD均來自上海源葉生物科技有限公司,本文所使用的溶劑均為分析純或更高,實驗用水為超純水(25 ℃時電阻率為18.2 MΩ·cm)。
1.2 實驗方法
1.2.1 相溶解度法
準確配制50 mL濃度為0.1 mol/L的CD標準溶液待用。準確稱量6份質量相同的CPT于15 mL離心管中,每份質量為0.001 0 g,再分別向這6支離心管中加入一定體積CD儲液并用水稀釋至10.0 mL,使其中CD濃度分別達到0、2.0×10-4、4.0×10-4、6.0×10-4、8.0×10-4和1.0×10-3mol/L。將上述樣品在室溫下用連續超聲30 min后在3 000 rpm條件下離心10 min,取上層清液通過0.22 μm微孔濾膜進行過濾,然后使用紫外-可見光譜分析。
1.2.2 傅里葉變換紅外光譜法
以DM-β-CD為例。將CPT、DM-β-CD、DM-β-CD/CPT物理混合物(摩爾比1∶1,研磨30 s)、DM-β-CD/CPT復合物(摩爾比1∶1,研磨30 min)干燥備用。分別取上述4種樣品與KBr按1∶100混合于瑪瑙研體中,在紅外光燈下混合研磨均勻后壓片,在波數400~4 000 cm-1范圍內檢測其紅外吸收。
1.2.3 分子模擬計算
α-CD(編號:BAJJAX)、β-CD(編號:ARUXIU)、γ-CD(編號:CYDXPL)的三維結構均來自于劍橋晶體數據庫。DM-β-CD、2HP-β-CD與6Glu-β-CD等β-CD衍生物結構是基于β-CD結構使用PyMol軟件自行繪制獲得的。CPT結構數據來自PubChem網站。CPT與所有CD分子在進行分子模擬計算前,均使用MMFF94方法[12]進行預處理。
本文使用Autodock 4.2軟件包進行主客體對接模擬[13]。主客體分子均依照Autodock系統程序默認值進行處理,未改變單鍵的自由度、非極性氫原子等。分子對接選擇在一個126 ?×126 ?×126 ?的立方體格子中進行,格子間隔為默認值0.375 ?,對接計算采用的是拉馬克遺傳算法(LGA)。為了使每次計算更為充分,以便獲得更為準確的計算結果,本文采用較大的運算參數以提高對接計算量,部分參數設置為:最大能量評估值(ga_num_evals)增加至2 500 000,種群數(ga_pop_size)提高至150,循環計算次數(ga_run)提高至300。
本文利用半經驗量子化學計算軟件MOPAC 2016(V17.279L)對分子對接所獲得的結果在半經驗水平進行量子化學計算[14],以便于探究復合物形成前后CPT與CD能量變化情況。分別采用PM6-D3H4和PM7方法進行計算,計算參數還包括XYZ、PRECISE、EF及GNOME=0.1,溫度為298.15 K。
復合物的復合能ΔEcomplexation依照下述公式計算[15-16]:
ΔEcomplexation=Ecomplex-(Ehost+Eguest) (1)
能量最低的分子對接結果隨后導入Schrodinger公司Maestro軟件包(2016.04)中,隨后使用Desmond分子動力學軟件[17]對其進行化學動力學模擬,進而評價分子對接結果的穩定性。
操作順序如下:首先將能量最低(即得分最高)的分子對接結果導入Maestro中,經過加氫和化學鍵鍵級糾錯后,再將其置于一個棱長為10 ?的立方體箱子中心,這個箱子中填充滿TIP3P模式水分子。由于CPT與CD分子均為電中性并且前述研究中所涉及的水溶液中并無緩沖鹽存在,因此這個體系中無需添加任何離子。使用Desmond默認參數進行體系弛豫過程和體系能量的最小化。分子動力學模擬采用NPT系綜,溫度采用Nose-Hoover耦合方法并設定為300 K,弛豫時間為1 ps,壓力采用Martyna-Tobias-Klein方法控制為1.013 25 bar,弛豫時間為2 ps,采用各向同性壓強耦合,模擬積分步長設置為5 fs。體系經過2 ns平衡后進行分子動力學模擬,時長為30 ns。
2 結果與討論
2.1 相溶解度法分析
圖2給出了通過監測CPT的λmax處(369.5 nm)吸光度所得到的6種CD存在時CPT的相溶解度曲線。由圖可見,在CD濃度為1×10-4~1×10-3mol/L范圍內,CPT溶解度S與CD濃度c之間存在良好的線性關系,線性方程及相關系數R2見表1。由圖2中曲線可以看出,當濃度為1.0×10-3mol/L時,DM-β-CD具有最大的增溶作用,而γ-CD的增溶作用幾乎不隨濃度變化,本文推測這可能是γ-CD空腔較大,結合客體能力不強導致。

表1 不同CD/CPT復合物的平衡常數Table 1 The stability constants of CD/CPT inclusion complexes
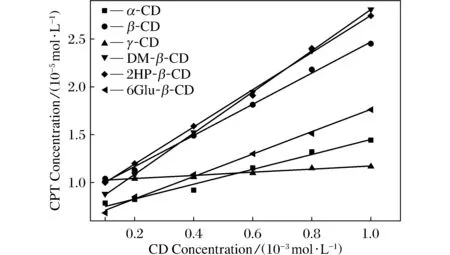
圖2 多種CD存在時CPT的相溶解度曲線Fig.2 Phase solubility curves of CPT in aqueous solution with the existence of various CDs
根據Higuchi和Connors[18]的分類,圖2中CPT與各種CD的相溶解度曲線均為AL型,表明CPT和CD之間均形成1∶1型復合物,根據公式K=斜率/(S0×(1-斜率))可計算出復合物的結合常數,其中S0為CPT在水中溶解度,斜率是各條擬合直線斜率。經實驗獲得CPT在水中溶解度S0為1.000 53×10-5mol·L-1,由此計算得到6種CD/CPT復合物的平衡常數K見表1,可以看出DM-β-CD的K值最大,具有最大的增溶作用。
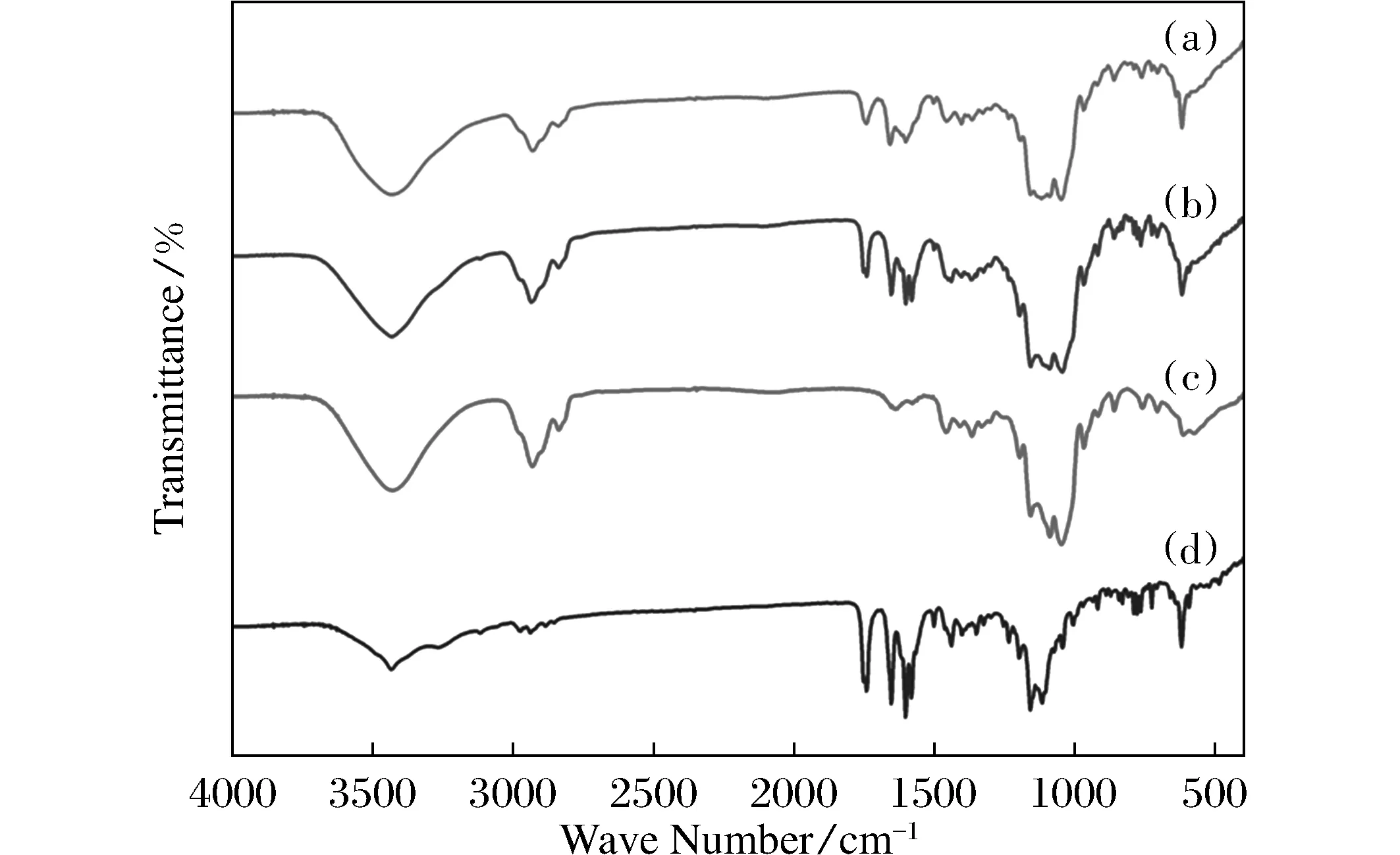
圖3 DM-β -CD/CPT復合物(A)、DM-β -CD/CPT物理混合物(B)、DM-β -CD(C)與CPT(D)的紅外光譜圖Fig.3 FT-IR spectra of DM-β -CD/CPT inclusion complex(A), DM-β -CD/CPT physical mixture(B), DM-β -CD(C) andCPT(D), respectively
2.2 紅外光譜分析
圖3為DM-β-CD/CPT復合物、DM-β-CD/CPT物理混合物、DM-β-CD與CPT紅外光譜圖。通過比對可以看出,1 741 cm-1處羰基吸收峰在物理混合物與復合物譜圖中均存在,但是在復合物譜圖中強度明顯下降,顯示復合物中此羰基與DM-β-CD存在相互作用;CPT的1 581 cm-1處吸收峰則在復合物譜圖中完全消失,說明CPT的芳香環骨架振動受到嚴重限制,推測是復合物中CPT復合在DM-β-CD空腔中所致。
2.3 分子模擬計算結果
本文采用分子對接推測DM-β-CD/CPT復合物的結構,300次分子對接后所得的能量最低對接結果示于圖4。從圖可知CPT分子沿著大口深入DM-β-CD空腔中,芳香環部分幾乎完全被包結在DM-β-CD疏水性空腔內,羥基、羰基等官能團位于DM-β-CD大口邊緣處,并且分別與DM-β-CD之間形成距離分別為2.3、2.6與3.0 ?的3個氫鍵。
為了考查分子對接結果的可靠性, 本文還通過半經驗計算探究了復合物形成前后主客體分子能量變化情況, 所得結果列于表2。 表2所示的ΔEcomplexation值均小于0可知復合物能量小于主客體能量之和, 即CPT與DM-β-CD復合在熱力學上是自發的。無論采用PM6-D3H4還是PM7方法計算, 都可獲得相近的結果,說明DM-β-CD/CPT復合物具有很好的熱力學穩定性, 驗證了分子對接結果的可靠性。

表2 DM-β -CD、CPT與DM-β -CD/CPT的半經驗計算結果Table 2 Semiempirical calculation results of DM-β -CD, CPT and DM-β -CD/CPT
本文還通過分子動力學模擬考查了分子對接結果的可靠性和復合物的動力學穩定性。圖5為30 ns時間內DM-β-CD/CPT復合物中主、客體相對初始位置RMSD隨時間的變化情況。由圖可見在分子動力學模擬開始后,體系迅速達到平衡,主、客體中原子位置的變化均在非常小的范圍內。在30 ns時間內,DM-β-CD與CPT的平均RMSD分別為1.772 1與0.423 8 ?,標準偏差(SD)均比較小,分別為0.271 3與0.051 93 ?,表明主客體在30 ns時間內主客體均未發生較大的位置改變與形變,說明復合物結構穩定,動力學穩定性強。
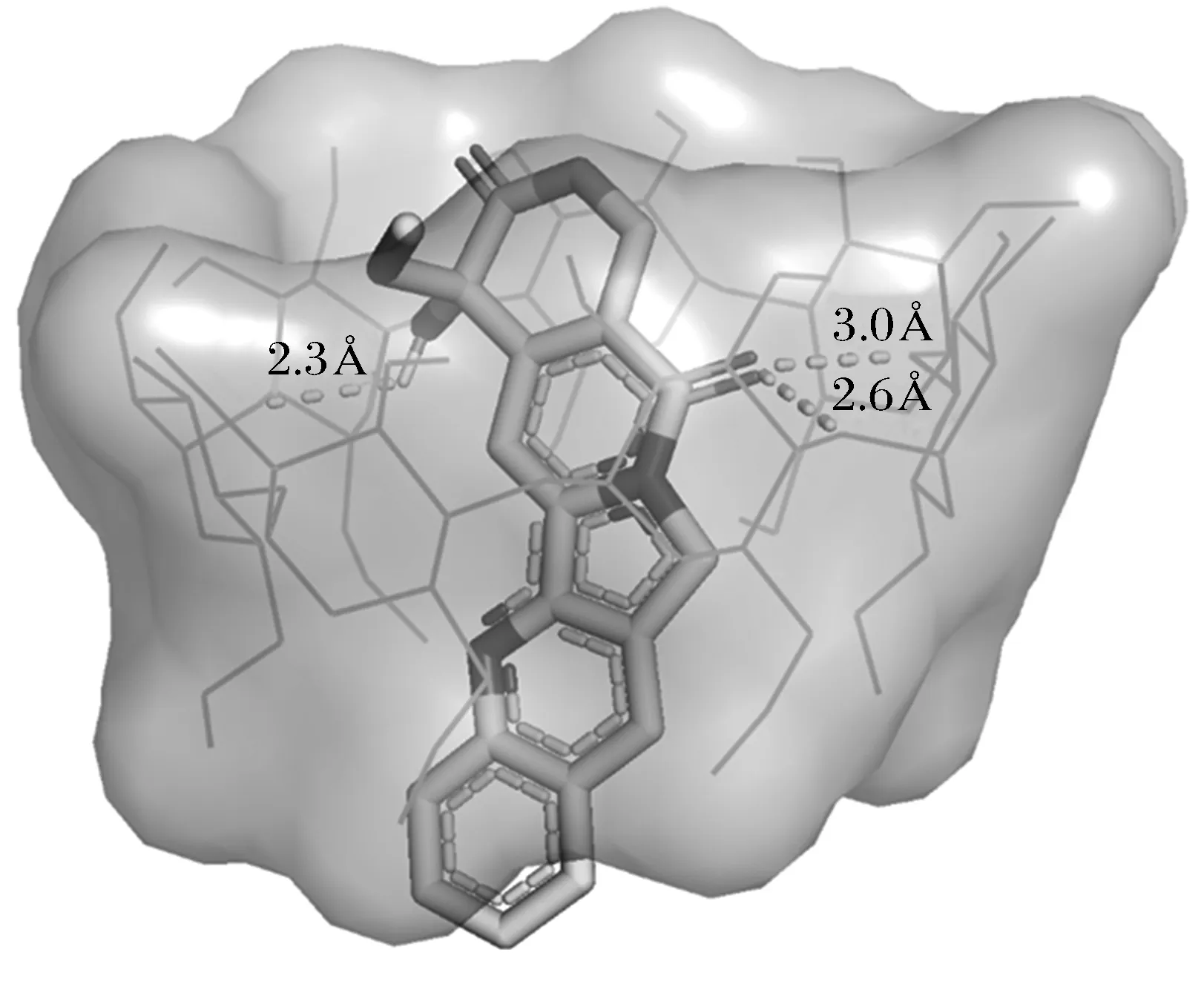
圖4 DM-β -CD與CPT最優分子對接結果
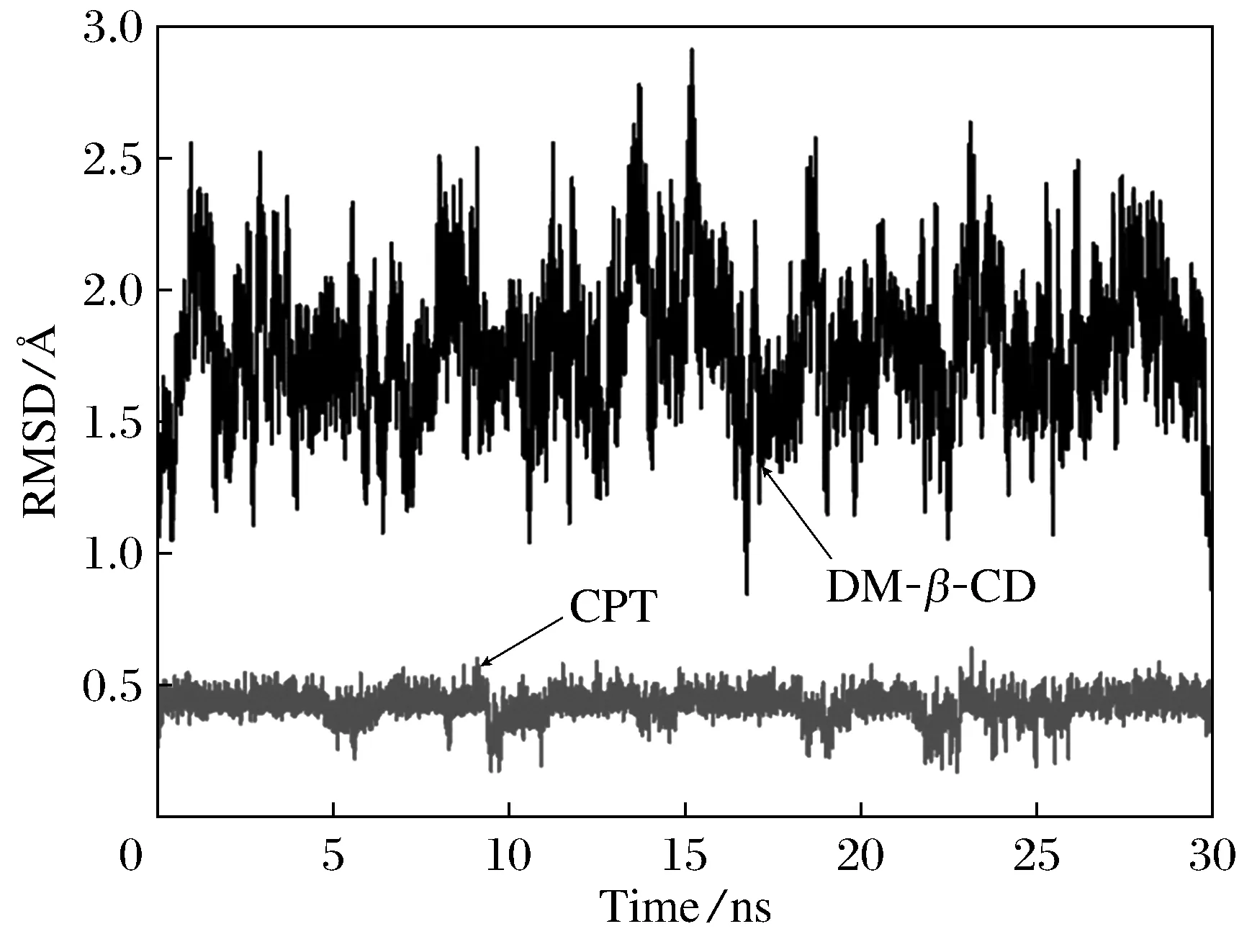
圖5 DM-β -CD/CPT復合物中DM-β -CD和CPT相對于初始結構的RMSD隨時間變化圖
3 結 論
本文采用相溶解度法研究了α-CD、β-CD、γ-CD、DM-β-CD、2HP-β-CD、6Glu-β-CD等6種CD對CPT的增溶,結果表明這些增溶劑均可以與CPT形成1∶1型復合物并具有一定的增溶能力,其中DM-β-CD具有最強的增溶作用。
本文還通過FT-IR法驗證了DM-β-CD/CPT復合物的存在與結構特點,并采用分子對接技術推測了復合物的可能結構,結果表明CPT通過進入DM-β-CD空腔并且與端口處氧原子之間形成氫鍵方式形成復合物。半經驗量子化學計算與分子動力學模擬表明DM-β-CD/CPT復合物具有很強的熱力學與動力學穩定性。
天然環糊精具有安全無毒、包結能力強的特點,作為藥物輔料廣泛應用于各種藥物制劑研究中。化學修飾可提高環糊精的水溶性并影響其空腔大小與性質,雖然DM-β-CD的空腔體積略小于β-CD,但是具有更好的包結能力,與藥物形成的復合物也具有更好的穩定性與水溶性。復合物中主客體之間存在明顯的疏水作用與氫鍵并具有良好的穩定性。本文研究結果表明,DM-β-CD是一種CPT的良好增溶劑,由于其毒性低[19]、增溶效果明顯、所形成的復合物穩定性強,在難溶性藥物制劑開發與臨床應用方面具有很好的潛力。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50