類風濕性嗜中性皮炎一例并文獻復習
2020-04-30 01:21:58俞婉婷劉澤虎
中國麻風皮膚病雜志 2020年4期
楊 珍 俞婉婷 劉澤虎 沈 宏
杭州市第三人民醫院皮膚科,杭州,310009
類風濕性嗜中性皮炎(rheumatoid neutrophilic dermatitis,RND)臨床較少見,易誤診。本文報道1例老年女性RND患者,其臨床皮損為多形性表現,根據生化和典型病理表現診斷,并進行相關文獻復習,有助于臨床醫生熟悉本病,避免誤診漏診。
1 臨床資料
患者,女,60歲,農民。因四肢紅斑、丘疹、結節痛1年余,加重伴水皰、膿皰1周于2019年5月30日至我院就診。1年余前無明顯誘因雙手足出現紅斑、丘疹,感疼痛,后皮疹逐漸增多,四肢關節伸側出現肥厚性斑塊、疣狀增生,多發結節,皮疹表面可見瘀點,多次就診當地醫院,具體治療不詳,效果不佳;1周前四肢原有皮疹及軀干正常皮膚上出現紅斑、環形紅斑及丘疹,環形紅斑上密集水皰,少許膿皰,綠豆至花生米大,水皰壁厚,不易破,后皮疹逐漸增多,自覺皮疹疼痛及頭痛,至我院進一步診治。患者確診“類風濕關節炎”3年余,近1年口服“甲潑尼龍片(美卓樂)8mg/d”,無明顯關節疼痛。家族中無類似疾病史。
體檢:一般情況好,雙手部分指關節輕度變形,無紅腫壓痛,無活動受限。余各系統檢查未見明顯異常。皮膚科檢查:軀干、四肢(以四肢為主)見大小不一、形狀不規則多發紅斑、環形紅斑和斑塊,其上及正常皮膚可見米粒至花生米大丘疹、水皰及膿皰四肢關節伸側可見疣狀斑塊、結節,部分紅斑表面、水皰破潰后可見糜爛面,滲出不明顯,皮疹上可見淤點(圖1)。
實驗室檢查及影像學檢查相關陽性結果:凝血功能+D二聚體:活化部分凝血活酶時間20.5 sec↓、D-二聚體(定量)2.24 mg/L↑;抗核抗體:抗著絲點抗體(Cent B)陽性+、抗線粒體抗體M2型陽性++、抗著絲點抗體陽性+、抗線粒體抗體陽性++;免疫功能:補體C4 0.58 g/L↑;全血C反應蛋白20.4 mg/L↑;類風濕因子504 IU/mL;血沉41 mm/h;CCP 101 Ru/mL;人類白細胞抗原B27(HLA-B27)陰性;甲狀腺功能八項+腫瘤篩查:甲狀腺過氧化物酶抗體2381.4 IU/mL↑、糖類抗原72~410.02 U/mL↑、甲狀腺球蛋白1.05 ng/mL↓、甲狀腺球蛋白抗體278.0 IU/mL↑;行B超(甲狀腺)檢查提示:甲狀腺回聲改變,TI-RADS 0類;行CT(胸部)檢查提示:兩下肺少許炎癥性病變。抗心磷脂抗體:陰性;ANCA:陰性;行磁共振(顱腦)檢查提示:腦血管MRA及彌散成像未見明顯異常。皮損組織病理檢查(圖2):①背部水皰:表皮下大皰形成,真皮淺層密集中性粒細胞浸潤,可見核塵,紅細胞外溢;直接免疫熒光:IgG、IgM、IgA、C3均陰性;②右肘部疣狀斑塊:表皮顯著角化過度,表皮乳頭瘤樣增生,棘層增生肥厚,真皮纖維組織增生,血管擴張,內皮細胞腫脹,部分管腔內血栓形成,周圍淋巴細胞、漿細胞浸潤;③左膝部:表皮突變平消失,真皮彌漫中性粒細胞伴淋巴組織細胞浸潤,可見核塵,血管擴張,內皮細胞腫脹。三次病理均無血管炎表現。
診斷:類風濕性嗜中性皮炎。
治療:予口服甲波尼龍片8mg日3次,雷公藤多苷片20mg日2次,并予外用鹵米松/三氯生乳膏等治療。2周后新發水皰減少,但仍有新發皮損,疼痛好轉,部分紅斑較前變淡;復查類風濕因子、CCP、CRP、ESR均較前明顯下降。繼續鞏固治療4周后無明顯新發皮疹。2.5個月后美卓樂逐漸減量至8mg日2次,雷公藤多苷片(治療2個月)停用。目前當地醫院風濕科及皮膚科門診定期復診中。

圖1軀干四肢多發紅斑、丘疹及斑塊,關節伸側多發斑塊、結節,其上可見淤點,軀干四肢散在或聚集水皰、膿皰,部分呈環形
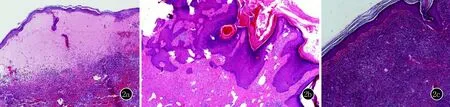
2a:表皮下大皰形成;2b:表皮角化過度,乳頭瘤樣增生,棘層增生;2c:表皮變薄,表皮突變平消失
圖2三次組織病理均顯示真皮彌漫性彌漫中性粒細胞伴淋巴組織細胞浸潤,可見核塵,血管擴張,血管內皮細胞腫脹,無血管炎改變(HE,×100)
2 討論
類風濕關節炎(rheumatoid arthritis,RA)是一種累及多關節、致關節畸形及功能喪失的全身性自身免疫性疾病,其亦可累及心、肺、神經系統和皮膚[1]。RA最常見的皮膚表現為類風濕結節、類風濕血管炎、壞疽性膿皮病等;與RA相關的不常見的皮膚病包括蕁麻疹、間質性肉芽腫性皮炎、匍行性回狀紅斑[2]、RND等。其中RND是一種少見的嚴重RA的皮膚表現,主要見于中年女性,多見于5年以上致殘性嚴重類風濕關節炎患者,也可發生于RA之前或其早期[3],皮損多與RA病情平行。皮損常對稱發生于四肢伸側、軀干及臀部,尤以腕部、手、指趾等關節伸側明顯[3,4],皮疹可表現為紅色、暗紅色丘疹、斑塊、結節、蕁麻疹樣風團或潰瘍,也可見水皰、膿皰或環形損害及淤點、瘀斑[5,6]。一般情況下無臨床癥狀,部分患者皮疹輕度瘙癢或自發疼痛或觸痛[4,7]。病理上以真皮內中性粒細胞浸潤為特征,可見明顯的白細胞破碎(核塵),血管內皮細胞腫脹,紅細胞外溢,但無血管炎和纖維蛋白沉積[5],DIF常陰性;類風濕因子多為高滴度陽性,本病由Ackerman在1978年首次報道[8]。
RND診斷需臨床表現、實驗室檢查結合組織病理。本病例有環形水皰,需與免疫性皰病相鑒別,如線狀IgA大皰性皮病(LABD),LABD是一種累及皮膚和黏膜的慢性獲得性自身免疫性疾病,表現為紅斑或外觀正常皮膚上周邊帶張力性水皰的環形皮損,病理見以中性粒細胞浸潤為主的表皮下水皰,DIF檢查見基底膜帶線狀IgA沉積;本例患者有軀干四肢有環形水皰,皮疹為累及黏膜,且病理DIF陰性,可排除;此外還可以排除皰疹樣皮炎、大皰性紅斑狼瘡、線狀IgA大皰性皮病、獲得性大皰性表皮松解癥。患者關節伸側疣狀斑塊、結節皮損,需與持久性隆起性紅斑(EED)鑒別,EED表現為關節伸側對稱性紫紅色斑塊和結節,病理上有白細胞碎裂性血管炎改變,而RND缺乏血管炎改變,故可予以鑒別。RND還需與Sweet綜合征、壞疽性膿皮病、Behcet綜合征等相鑒;RND和Sweet綜合征組織學表現相似,但Sweet綜合征常伴有發熱、乏力、關節痛、結膜炎等癥狀,外周血白細胞升高,面部及四肢的復發性疼痛性水腫性紅斑;而RND一般無全身癥狀。壞疽性膿皮病多表現為雙下肢對稱多發的大的壞死性疼痛性潰瘍,組織學表現為嗜中性粒細胞浸潤伴血管炎改變,而RND組織病理上無血管炎改變,因此可以與其鑒別。Behcet綜合征表現為口腔、外生殖器潰瘍與虹膜炎,但病理表現為血管炎或嗜中性血管炎癥反應,可鑒別。
RND發病機制清不清楚,免疫復合物的活化、細胞粘附和遷移及及趨化因子(如IL-6、IL-8)的釋放[9],被認為是RND可能的發病機制,特別是IL-8在皮膚內中性粒細胞的聚集中起重要作用[4,6],這就可以解釋使用托珠單抗(抗人IL-6受體抗體)治療類風濕關節炎時出現類風濕性中性粒細胞性皮炎[10]這一現象。有文獻曾指出膠原蛋白的早期改變,即纖維素樣變性也可能在RND的發病機制中起作用[1]。
RND治療一般采用糖皮質激素、雷公藤、羥氯喹[11]、氨苯砜[12]、環磷酰胺[13]、阿奇霉素[14,15]及外用激素[9]等。依那西普(抗TNF受體單抗)治療大皰性RND效果良好[15,16],環磷酰胺對頑固性損害有效。非甾體類抗炎藥無效[1]。大部分患者的皮疹可隨RA的病情改善而消退[1,9],部分患者可自行緩解。
本例患者RA病史只有3年,且無明顯關節畸形改變,可能與長期口服激素有關;患者RF滴度較高,出現紅斑、環形紅斑、斑塊、結節、水皰、膿皰及瘀點等多形性皮損,考慮與RA病情活動及激素劑量較低有關。患者皮損多形性較少見,未見以往相關文獻報道;患者多次不同皮損活檢病理除表皮表現不同外,真皮細胞浸潤及血管表現均一致,為彌漫中性粒細胞浸潤,無血管炎改變及纖維蛋白沉積,DIF無補體沉積;患者入院后口服復方對乙酰氨基酚片(散利痛),皮疹及疼痛均無好轉,證實非甾體抗炎藥無效。給予甲潑尼龍片(美卓樂)加量及加用雷公藤多苷片抗炎抑制免疫治療后,起始1周患者仍有新發水皰,2周后復查CRP、RF、ESR等指標明顯下降,治療約2個月皮損穩定,無明顯新發水皰,提示皮疹好轉延后于RA病情(相關炎癥指標)好轉,但有可能只是個例。
