腹膜后Castleman病CT及MR影像特征與鑒別診斷
2020-03-25 13:24:08杜亮崔鳳熊發奎
浙江臨床醫學 2020年2期
關鍵詞:信號
杜亮 崔鳳 熊發奎
Castleman病(Castleman’s disease,CD)是一種少見的慢性淋巴組織增生性疾病,1954年由Benjamin Castleman首次報道,1972年由keller正式命名。該病主要發生于縱隔,腹膜后間隙內發病率較低[1]。因CD影像表現多樣,常將其誤診。作者收集近3年經手術或病理證實的4例CD,分析其影像特點,旨在提高影像醫生對該病的認識及診斷能力。
1 資料與方法
1.1 一般資料 回顧性收集2016年1月至2018年12月經手術或病理證實的4例腹膜后Castleman 病患者,其中男1例,女3例;年齡24~58歲,平均43歲;2例病灶位于左側腎門旁,2例位于腸系膜根部。臨床表現:1例以右下腹疼痛6h來診,擬急性闌尾炎收治,以往有特發性嗜酸性粒細胞增多癥,長期服用激素類藥物;1例以陣發性腹部隱痛來診,觸診左上腹部質韌腫塊,移動度可;2例體檢發現病灶,平素無任何癥狀,實驗室指標均正常。
1.2 檢查方法 采用飛利浦 256iCT掃描,對比劑為優維顯80~120ml,采用 CT專用高壓注射器,注射流率2.5~3.0ml/s,動脈期30s,靜脈期75s,平衡期3~5min,所有圖像應用MPR、VR、CPR、CTA等圖像后處理進行觀察,分析其影像學特征;MRI掃描采 用 GE Discovery 3.0T 超 導 MRI,T1WI(TR196ms,TE2.6ms)、T2WI(TR 1200ms,TE 60ms),層厚 8mm,層間距10mm,對比劑為釓噴酸葡胺注射液0.2mmol/kg進行靜脈團注(2ml/s),層厚5mm,延遲時間分別為動脈期15s,門脈期35s,平衡期1min。
2 結果
2.1 影像表現 本組4例病灶,1例位于后腹膜左腎動靜脈間,1例位于左側腎門旁(見圖1),2例位于腸系膜根部,長徑分別約為9.5cm、2.7cm、3.6cm、3.3cm,病灶形態以橢圓形、類圓形及結節狀為主,部分可見輕度分葉狀,邊界尚清。(1)CT平掃:病例1病灶密度不均勻,可見斑點狀鈣化及散在斑片狀稍低密度影,CT值約26~43HU,病例2~4病灶密度較均勻,CT值分別約34~40HU、42HU及37~43HU,病例3病灶內亦見少許點狀鈣化。動態增強:病例1見腹主動脈發出供血小動脈,病灶呈不均勻漸進性強化,門脈及延遲期強化逐漸均勻,最大CT值約85HU;病例2~4動脈期病灶明顯強化,最大CT值分別約148HU、155HU、140HU,隨后強化程度稍減退、呈延遲強化表現,近似腹主動脈強化特點。病例2及病例4動脈期可見裂隙樣欠強化區(見圖2),靜脈及延遲期裂隙影逐漸填充;另外,2例病灶周圍均可見小結節影(見圖3),與主病灶同步強化。(2)MRI平掃:腫塊信號不均勻,呈稍長T1稍長T2信號為主,內見斑片狀短T2信號,DWI序列為不均性高信號(見圖4),增強掃描動脈期病灶中央見少許斑片狀強化(見圖5),動態各期病灶強化范圍增大、呈進行性強化表現(見圖6),邊界尚清,左腎上腺及腎動靜脈受壓移位。T2WI冠狀位重建:不均勻稍高信號腫塊內伴裂隙樣低信號影(見圖7)。CTA:可見明顯供血動脈來源于腹主動脈(見圖8)。
2.2 手術與病理 病例1腫瘤表面可見大量迂曲的血管,與腎靜脈粘連,鈍性分離并完整切除,病理:淋巴組織增生,濾泡間大量漿細胞浸潤,間質彌漫纖維化、玻璃樣變及血管增生。免疫組化:血管內皮CD31、CD34(+),濾泡內淋巴細胞 CD20、PAX5(+),濾泡間淋巴細胞 CD3 、CD5、Bcl-2(+),CD21示FDC網,Ki-67濾泡內約70%。病理診斷:混合型,Castleman 病(巨淋巴結增生癥)(見圖9)。病例2濾泡間區大量小血管及淋巴組織增生,部分套區淋巴組織呈同心圓樣排列,間質散在少量漿細胞,符合透明血管型Castleman 病。免疫組化結果: CD20(+),CD21(+),CD34 血管(+),CD 10(局灶 +),Ki-67(20%+)。病例3淋巴組織增生,以透明血管型為主。免疫組化:CD20(+),CD21(+),CD34(生發中心濾泡樹狀突細胞),FVⅢ(+)。病例4病灶散在分布的淋巴濾泡,濾泡間可見明顯的小血管增生,存在異常生發中心形成和明顯的玻璃樣變性,內部見纖維組織及炎性細胞,提示淋巴增生性病變,透明血管型Castleman。
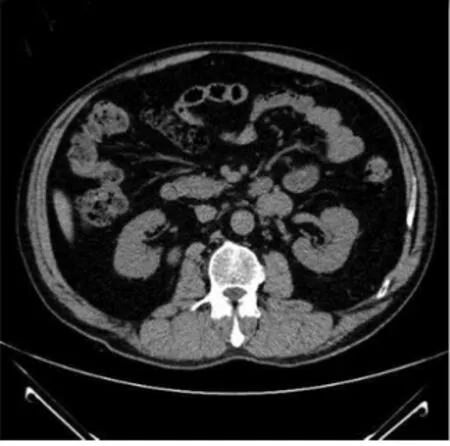
圖1 CT平掃左側腎門軟組織占位
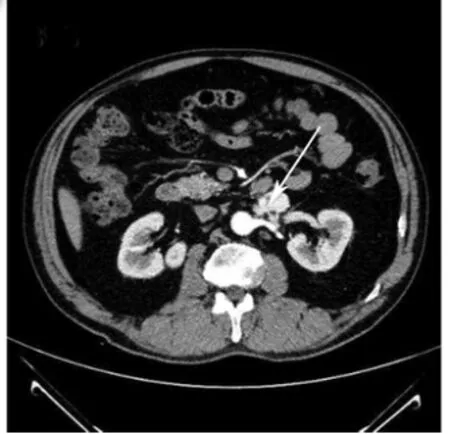
圖2 動脈期明顯強化,中央見裂隙(箭頭)
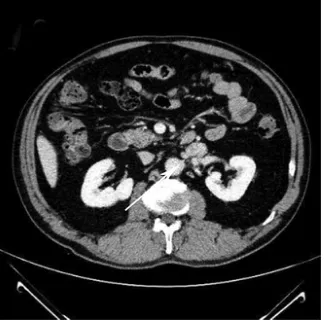
圖3 靜脈期病灶周圍衛星灶(箭頭)
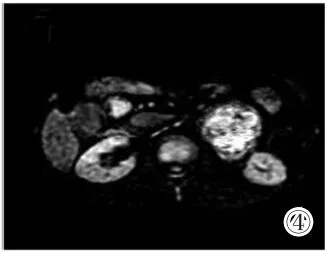
圖4 DWI不均勻高信號。
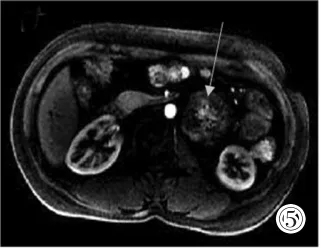
圖5 T1WI增強動脈期左腎前占位(箭頭),中央輕度強化。
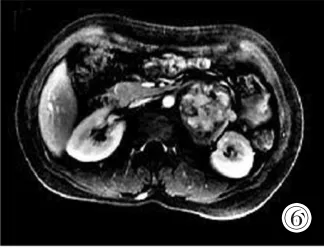
圖6 T1WI延遲期強化范圍增大,伴裂隙樣無強化區。
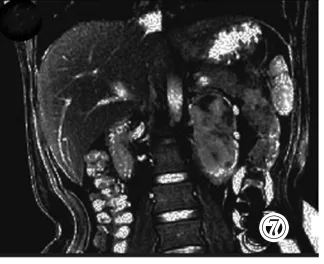
圖7 T2WI冠狀位抑脂不均勻稍高信號影伴低信號裂隙影。
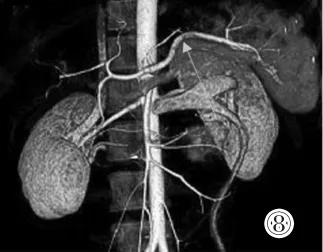
圖8 CTA示腫塊(箭頭)由腹主動脈發出小動脈供血。
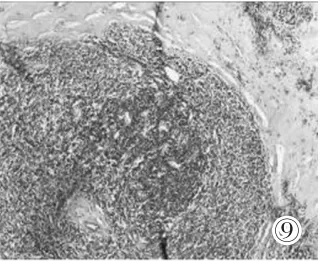
圖9 病理(HE×100):淋巴組織增生,濾泡間大量漿細胞浸潤,間質彌漫纖維化、玻璃樣變及血管增生。
3 討論
3.1 病理及分類 Castleman’s disease簡稱CD,即血管濾泡性淋巴增生癥或巨淋巴結增生癥,白細胞介素-6(IL-6)異常表達和人皰疹病毒8(HHV-8)感染為公認的發病原因[2]。參照Frizzera的診斷標準[3],CD病理上分3型,透明血管型、漿細胞型和混合型。(1)透明血管型相對多見,增大的濾泡樣淋巴結構,可見小血管進入,鏡下見淋巴濾泡增生及濾泡間玻璃樣變的小血管;免疫組化:CD20(+),CD3(+),CD21(+),CD34血管(+),本組2~4病例淋巴濾泡增生,不同程度玻璃樣變性,為透明血管型的典型病理表現。(2)漿細胞型少見,淋巴濾泡樣增生,濾泡間可見各級漿細胞增生,鏡下以濾泡間漿細胞浸潤為主;免疫組化:CD20濾泡(+),CD3濾泡間(+),CD138、CD38濾泡間(+),Ki 67陽性細胞數3%,Bcl 2濾泡間(+),CD21濾泡(+),CD34血管(+),本組無漿細胞型病例。(3)混合型有透明血管型和漿細胞型的共同特點,病例1增生濾泡間見大量漿細胞侵潤,符合混合型病理表現。
3.2 臨床表現 臨床上將CD分為局灶型和多中心型,局灶型病灶多無癥狀,多中心型患者通常表現為全身多部位淋巴結腫大,同時可出現發熱、乏力、貧血,肝脾腫大等全身表現,腹膜后CD可合并皮疹、天皰瘡,臨床上被稱為副腫瘤性天皰瘡[4]。局灶型CD好發于縱隔,手術切除預后良好,有明確供血動脈者,術中應結扎腫瘤的滋養血管[5];多中心型CD預后較差,多采用放射治療或化學治療。Hill等[6]報道CD具有不同亞型而表現多樣,并發現CD具有臨床模擬淋巴瘤的全身B癥狀(發熱、盜汗、消瘦及皮膚痛癢)和血液紊亂的多灶腺病,其研究結果中3例罕見患者均先后出現全身B癥狀,并發展成為不同類型的淋巴瘤,故認為HHV-8相關(漿細胞)CD患者具有發生HHV-8陽性的漿細胞性淋巴瘤的獨特風險,積液和/或腹水可能預示淋巴瘤的發展。
3.3 影像表現 CD在影像上具有典型特征,該病沿著淋巴鏈分布,透明血管型患者多為一孤立性軟組織腫塊,邊界光整,其內可見斑點狀、樹枝狀鈣化,平掃CT值約30~60HU,動脈期呈明顯均勻或不均勻強化,CT值增幅>50HU,靜脈及延遲期呈持續強化,強化程度略低于動脈期,該強化方式與主動脈強化相似,當腫塊較大時病灶中心出現裂隙狀低密度灶,一般以直徑5cm為參考標準,病理上認為中心低密度是小血管透明樣變和纖維化[7],增強掃描后中央低密度范圍逐漸縮小,呈外周向中央填充表現;李博等[8]報道腹膜后透明血管型CD病灶邊緣薄環形強化,病灶周圍可見供血血管影,本組有1例較大病灶內出現供血動脈,但4例均未出現環形強化表現;據多篇文獻報道病灶周圍可有衛星灶出現,本組有2例可見結節樣類似病灶。漿細胞型患者CT表現為一組或多組增大淋巴結,病灶較小,密度均勻,CT增強掃描病灶動脈期無強化或輕度強化,靜脈期持續中度強化,強化程度與肌組織相似,這可能與濾泡間大量漿細胞浸潤,血管增生稀少有關。MRI:T1WI呈等低信號或低信號、T2WI呈高信號,DWI序列呈高信號,當出現鈣化、瘢痕及纖維化成分時,T1WI、T2WI均出現低信號,增強掃描時其強化方式同CT增強,但對于顯示供血動脈更有優勢。
3.4 鑒別診斷 (1)副神經節瘤:多為實性或囊實性腫塊,可見出血及液平,強化明顯,多數可見流空血管;透明血管型CD典型者可見樹枝狀鈣化,囊變少見,較大病灶內出現瘢痕纖維組織,腫塊邊緣衛星結節有助于CD的診斷。(2)神經鞘瘤:多沿神經或大血管走行的囊實性腫塊,易囊變為典型特點,多呈啞鈴狀或鼠尾狀生長,T2WI信號不均勻,多發小環、漸進性延遲強化是其特征;而漿細胞型CD亦呈漸進性強化,但沿淋巴鏈多灶分布,臨床上常出現腹痛、腹瀉、消瘦、貧血及淺表淋巴結腫大等癥狀。(3)淋巴瘤:與漿細胞型CD鑒別困難,無痛性淋巴結腫大,肝脾增大,有發熱、瘙癢、盜汗及消瘦的癥狀,部分癥狀與漿細胞型CD類似;且淋巴瘤為多發實性病灶,可融合呈團,并包繞鄰近的血管,即出現“血管漂浮征”,該征象有助于鑒別CD。(4)平滑肌肉瘤:腹膜后間隙罕見的惡性腫瘤,壞死多見,無鈣化,腫塊浸潤性生長,周圍血管組織易受侵,增強實性成分呈延遲強化,動脈期可見典型的瘤內血管影,該病需與混合型CD鑒別,另外平滑肌肉瘤多見種植和血行轉移,而非淋巴結轉移。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
