利用CRISPR/Cas9系統敲除大腸桿菌tnaA基因
2019-12-19 01:51:00何京樺樂科易
生物學雜志 2019年6期
何京樺, 樂科易
(復旦大學 生命科學學院, 上海 200433)
L-色氨酸在人和動物體內非常重要,同時在醫藥、食品和飼料添加劑等方面具有廣泛的用途[1-4]。大腸桿菌生長迅速、遺傳背景清晰,被認為是生產L-色氨酸的主要菌株。大腸桿菌的tnaA基因編碼色氨酸酶(tryptophanase),該酶在正常情況下可以將L-色氨酸降解成丙酮酸、吲哚和氨,從而使碳源進入TCA循環,阻礙L-色氨酸的積累[5-6]。tnaA基因的失活有利于大腸桿菌積累更多的L-色氨酸[7]。
成簇、規則間隔的短回文重復序列(clustered, regularly interspaced, short palindromic repeat,CRISPR)是細菌在長期進化過程中產生的抵抗外源基因入侵的適應性免疫系統[8-11]。CRISPR-Cas9系統為RNA介導的基因組編輯提供了強大的工具,并已廣泛應用于包括原核生物和真核生物在內的多種生物中[12-14]。在該系統中,CRISPR RNA(crRNA)通過與轉錄激活crRNA(trans-activating RNA,tracrRNA)結合形成tracrRNA/crRNA復合物,引導Cas9核酸內酶在目的DNA序列生成雙鏈斷裂(double-strand breaks,DSBs)[15-16]。gRNA與靶DNA序列中長約20 bp的片段互補配對,靶序列末端須含有PAM序列(5′-NGG)[17]。
本研究以E.coliw3110-Δ為出發菌株,利用CRISPR/Cas9基因編輯技術對其tnaA基因進行敲除,篩選出tnaA基因缺失菌株,減少色氨酸的降解,從而為L-色氨酸高產菌株的構建奠定基礎。
1 材料與方法
1.1 菌株與質粒
E.coliw3110-Δ由本實驗室保存,特征是酪氨酸缺陷型,適合于芳香族氨基酸發酵。pCas和pTargetF質粒通過Add gene載體庫(http://www.addgene.org/crispr-cas)獲得,編號分別為62225和62226。pTargetF-tnaA質粒由本研究構建。
1.2 主要試劑
DNA產物純化試劑盒(TIAN quick Mini Purification Kit)購于天根生化科技(北京)有限公司。DNA聚合酶(Prime STAR?Max DNA Polymerase)和限制性內切酶DpnI均購于寶生物工程(大連)有限公司。Gibson組裝克隆試劑盒(Gibson Assemly Cloning Kit)購自NEB(北京)有限公司。
1.3 gRNA質粒的構建
通過http://www.rgenome.net/cas-offinder/設計tnaA基因的gRNA。將設計出的20 bp序列加入引物gRNAR中。用一步等溫Gibson組裝法利用Prime STAR?Max DNA 聚合酶將pTargetF、上下游引物gRNA F、gRNA R進行組裝[18]。將獲得的產物用DpnI酶切,以去除多余的pTargetF。獲得的產物轉化到E.coliDH5α感受態細胞中,小提質粒,用測序引物gRNAP F/gRNAP R測序鑒定。將構建好的質粒命名為pTargetF-tnaA。本研究所用引物和gRNA寡核苷酸序列見表1。

表1 gRNA和引物
1.4 tnaA基因同源修復供體 DNA的構建
以E.coliw3110-Δ基因組DNA為模板,tnaA L F、tnaA L R為引物,PCR擴增獲得片段1,以tnaA R F、tnaA R R為引物,PCR擴增獲得片段2。以AmlF和AmlR為引物,將片段1和片段2用重疊延伸PCR法獲得同源修復供體DNA。
1.5 感受態細胞的制備及細菌的轉化
將pCas質粒轉化到E.coliw3110-Δ感受態細胞中,轉化產物涂布到含有卡那霉素(50 μg/mL)的LB瓊脂上(30 ℃培養),挑選獲得含有pCas質粒的E.coliw3110-Δ。將含有pCas質粒的E.coliw3110-Δ按照文獻[14]描述的方法制備感受態細胞。當感受態細胞生長至其OD600為0.1時,加入終濃度為10 mmol/L的阿拉伯糖,以誘導λ-Red介導的同源重組[19]。當OD600=0.6時收集菌體,將100 ng gRNA質粒pTargetF-tnaA和500 ng供體DNA加入到約100 μL含pCas質粒的E.coliw3110-Δ感受態細胞中,進行電轉(2 mm cuvette, 2.5 kV)。電轉后立即在1 mL LB液體培養基中懸浮細胞,30 ℃復蘇1 h,涂布至含有卡那霉素(50 μg/mL)和壯觀霉素(50 μg/mL)的LB瓊脂培養基上。30 ℃過夜培養,用引物tnaAP F和tnaAP R對轉化子進行PCR檢測,并進行測序鑒定,將測序結果和原基因序列進行對比,檢測tnaA基因是否敲除成功。
1.6 pTargetF-tnaA和pCas的消除
將鑒定正確的含有pCas和pTargetF-tnaA的克隆接種于含有卡那霉素(50 μg/mL)和IPTG(異丙基-β-D-硫代半乳糖苷,0.5 mmol/L)的2 mL LB培養基中,培養8到16 h后涂布到含有卡那霉素(50 μg/mL)的LB瓊脂上。然后以克隆對壯觀霉素(50 μg/mL)的敏感性確認pTargetF-tnaA已經被消除。將克隆在43 ℃培養過夜以消除pCas質粒[20]。
1.7 敲除效率的優化及檢測
為提高該敲除系統對大腸桿菌tnaA基因的敲除效率,在100 μL感受態細胞中,將pTargetF-tnaA質粒加入量由普遍采用的100 ng提高到300 ng, 供體DNA片段加入量由普遍采用的400 ng提高到1.5 μg進行轉化,用引物tnaAP F和tnaAP R對轉化子進行PCR鑒定和測序鑒定,計算該系統對tnaA的敲除效率。
2 結果與分析
2.1 gRNA的構建
用一步等溫Gibson組裝法利用Prime STAR?Max DNA 聚合酶將pTargetF質粒、上下游引物進行組裝。獲得的產物轉化到E.coliDH5α感受態細胞中,小提質粒,將提取的質粒pTargetF-tnaA送檢測序,測序結果正確,可用于下一步試驗。
2.2 tnaA基因同源修復供體 DNA的構建
分別以tnaA L F/R 和tnaARF/R為引物,PCR擴增獲得了片段1和片段2,以AmlF和AmlR為引物,利用片段1和片段2用重疊延伸PCR法獲得了同源修復供體 DNA。
2.3 tnaA基因的敲除及敲除效率的檢測
pCas質粒成功轉入E.coliw3110-Δ中。pTargetF-tnaA質粒和供體DNA成功共轉入含有pCas質粒的E.coliw3110-Δ中。用橫跨tnaA基因同源修復供體DNA的鑒定引物tnaAP F和tnaAP R擴增目的條帶來鑒定tnaA基因的敲除情況。結果(圖1)顯示,tnaA基因沒被敲除修復的菌,PCR顯示為1150 bp的條帶,反之出現750 bp的目的條帶。測序結果與設計的同源臂序列是一致的,表明敲除后的tnaA基因在人工設計的供體序列下發生了同源修復。挑取12個克隆進行PCR鑒定和測序鑒定,9個克隆敲除成功,該敲除系統對大腸桿菌tnaA的敲除效率為75%。以上結果均表明,本研究構建的CRISPR/Cas9系統在人工同源修復下,可對大腸桿菌tnaA基因進行高效敲除,圖2是敲除模式示意圖。
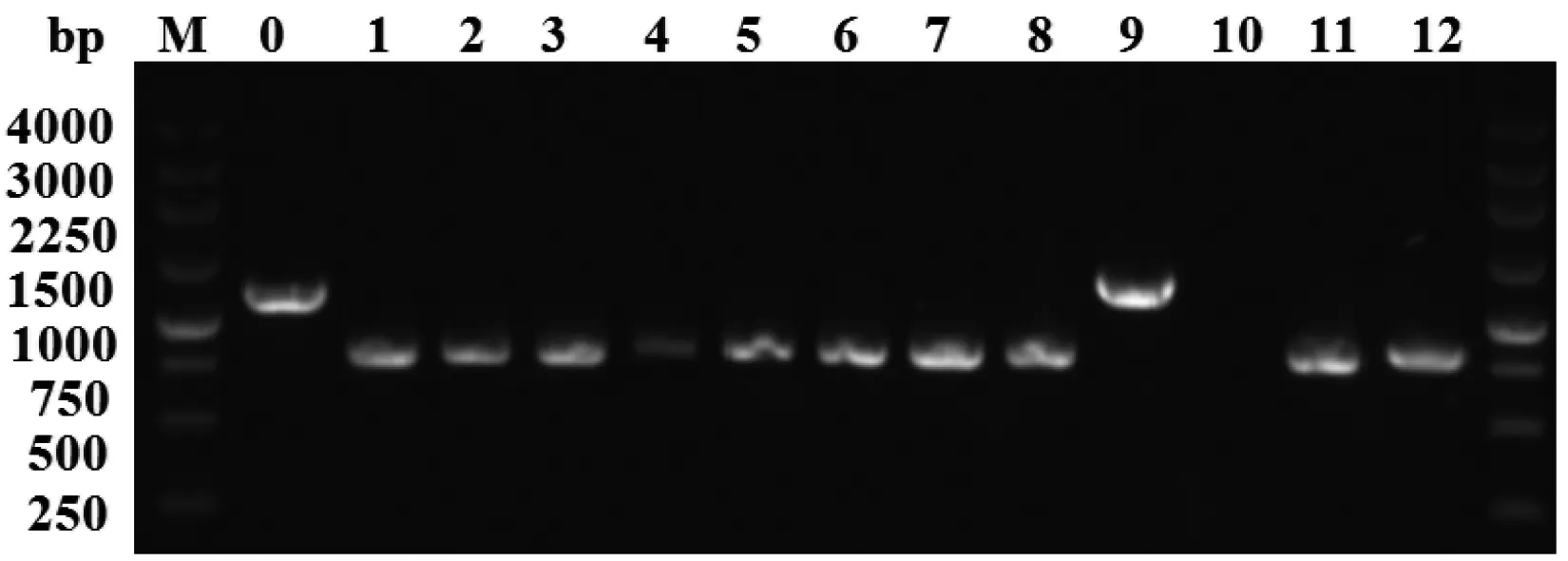
M:相對分子質量;0:原始菌的PCR產物;泳道4、9、10:未敲除成功的PCR產物,大小約為1150 bp;泳道1、2、3、5、6、7、8、11和12:敲除tnaA后的PCR產物,大小約為750 bp
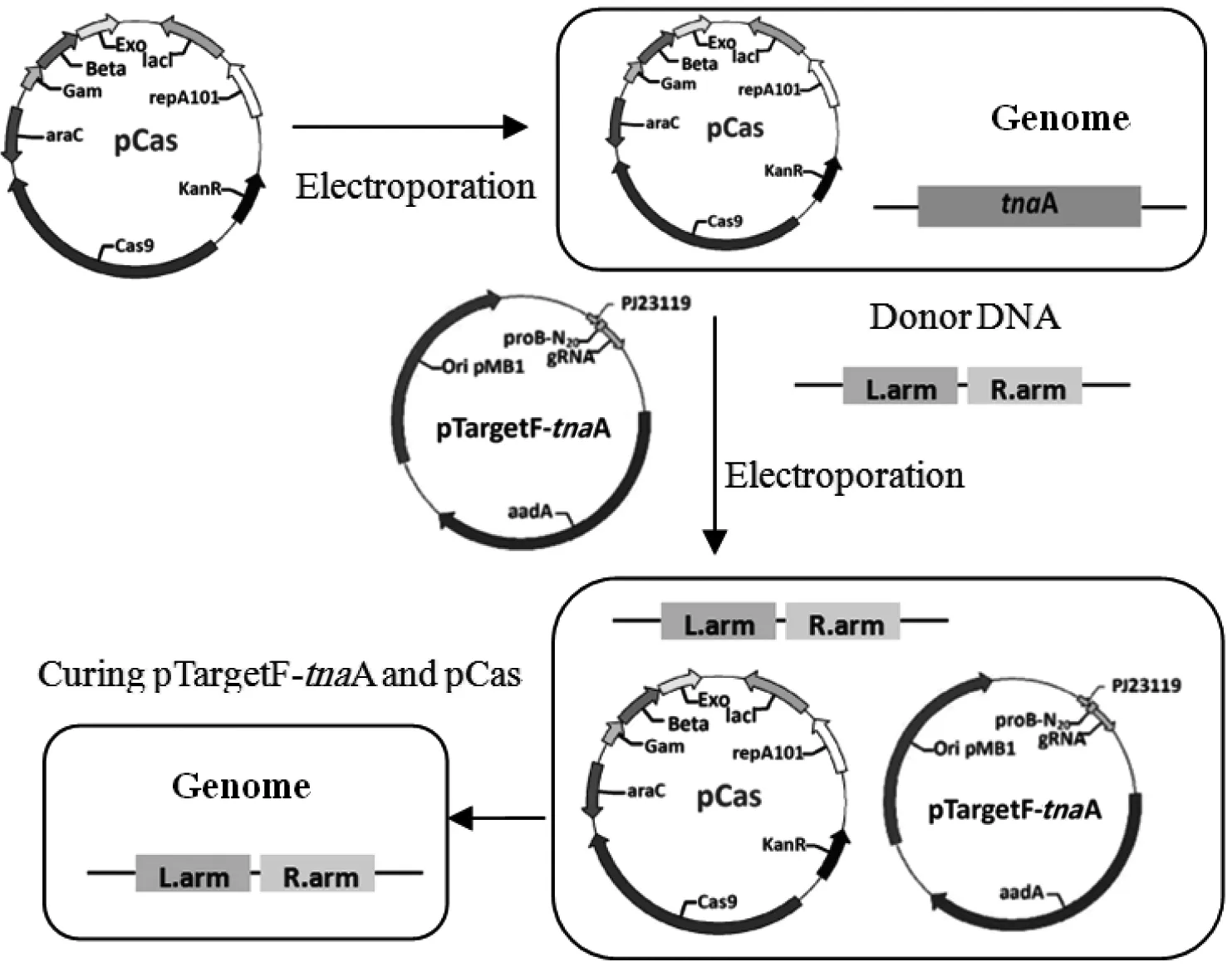
圖2 利用CRISPR/Cas9系統進行基因敲除示意圖
3 討論與結論
CRISPR-Cas9是細菌以及古細菌在長期進化過程中形成的一種適應性免疫機制,用以對抗入侵的病毒及外源DNA。CRISPR-Cas9基因編輯技術,則是對目的基因進行特定的DNA修飾的技術,該技術已應用于細菌、真菌、斑馬魚以及人類細胞的基因編輯。
在該技術中,一般是將CRISPR-Cas9技術與同源重組技術結合起來,構建含Cas9、sgRNA和Donor序列的重組載體,對目標基因進行有效準確的編輯。當然該技術也在不斷地被改進優化, Jiang等人在Cas9質粒構建時引入了Red重組系統,供體DNA序列不需要克隆到載體上,而是直接與質粒共轉化入宿主菌,使得該技術可以對多個基因同時進行高效的敲除[14]。
為了避免雙鏈斷裂DNA被同源修復后,gRNA/Cas9復合體對其重復切割,Wang等人采用了兩步法策略,第一步使用CRISPR/Cas9系統用20 bp人工序列替換了原始間隔序列,以避免gRNA/Cas9復合體的重復切割;第二步使用第二個gRNA識別引入的人工序列進行基因突變,同時人工序列被還原為原始序列,從而只引入一個點突變,且不會改變PAM或不會在基因組中插入額外的沉默突變[20]。
本研究在構建質粒時采用了Gibson等溫一步拼接法,該方法是目前最常用的新興克隆方法之一, 能在5′核酸外切酶、DNA聚合酶和DNA連接酶的協同作用下直接組裝多個重疊DNA分子,這種組裝方法可以無縫地構建基因, 是一種有用的分子工程工具。
本研究成功構建了大腸桿菌tnaA基因CRISPR/Cas9敲除系統,敲除效率75%。該系統將來也可應用于大腸桿菌其他基因的敲除,為構建L-色氨酸高產大腸桿菌提供有效的基因敲除工具。
