應用不同基因檢測方法分析假肥大型肌營養(yǎng)不良基因的變異特點
2019-12-06 08:45:26鄧艷春
中風與神經(jīng)疾病雜志 2019年11期
康 娟,鄧艷春
假肥大型肌營養(yǎng)不良癥(pseudohypertrophy muscular dystrophy)是最常見的遺傳性肌肉疾病,包括杜興型肌營養(yǎng)不良癥(duchenne muscular dystrophy,DMD)和貝克型肌營養(yǎng)不良癥(becker muscular dystrophy,BMD),二者均是由編碼抗肌萎縮蛋白(dystrophin,也稱DMD基因)基因突變所致的X-連鎖隱性遺傳病[1,2]。假肥大型肌營養(yǎng)不良癥患者由于抗肌萎縮蛋白缺乏導致了骨骼肌細胞膜缺陷,細胞內(nèi)的肌酸激酶等外漏,肌細胞壞死、脂肪組織和纖維結(jié)締組織增生[3]。臨床表現(xiàn)為骨骼肌的進行性萎縮、無力、腓腸肌假性肥大。其中DMD的發(fā)病率約占活產(chǎn)男嬰的1/3500[2]。此型患者通常于4~5歲表現(xiàn)出雙下肢運動乏力,呈進行性加重,8~12歲喪失行走能力,20歲左右死于呼吸衰竭或心力衰竭;BMD發(fā)病率約為1/12000[4],癥狀較輕,部分患者從16歲喪失行走能力,少數(shù)到50歲、60歲才出現(xiàn)臨床癥狀,病情嚴重程度不等[5]。目前國際上關于DMD/BMD的基因治療取得突破性進展,如通過輸注反義寡核苷酸誘導外顯子跳躍而糾正讀框移位,或在翻譯過程中抑制無義突變等方法[6~9],盡早明確臨床DMD/BMD診斷,為及時進行基因治療及綜合治療提供條件。目前臨床基因檢測方法較多,合理選擇檢測方法,利于高效、經(jīng)濟、準確為臨床提供驗證,DMD/BMD的基因診斷多采用多重連接探針擴增技術 (multiplex ligation-dependent probe amplification,MLPA),快速分析患者DMD/BMD基因缺失突變/重復突變,外顯組測序 (Exome sequencing) 補充點突變及拷貝數(shù)檢測,Sanger測序可用于點突變驗證[10~12]。本研究,我們合理選擇上述方法,對10例 DMD/BMD患者進行了基因突變分析。
1 對象與方法
1.1 對象 我們通過收集2016~2019年我科10例臨床診斷DMD/BMD患者的臨床資料,知情同意后,選擇性應用MLPA、外顯組測序及Sanger測序技術,從基因水平驗證臨床診斷。臨床納入標準:(1)臨床表現(xiàn)有進行性肌無力,以肢體近端為著,腓腸肌肥大,查體有Gower征陽性;(2)心肌酶譜化驗:肌酸激酶升高,正常值的數(shù)十倍及以上,并排除心臟和肝臟疾病;(3)肌電圖或肌肉活檢提示肌源性損害。
1.2 方法
1.2.1 基因組DNA提取 在知情同意的前提下,采集患者外周靜脈血2 ml于EDTA抗凝管中。使用BloodGen Midi Kit (CWBIO,中國) 試劑盒按照說明書進行提取患者全基因組DNA。
1.2.2 MLPA分析 SALSA MLPA探針組 P034-B2/P035-B1 DMD試劑盒購自荷蘭MRC-Holland公司 。用 TE溶液稀釋DNA樣本至 40 ng/μl,取 5 μl DNA樣本,依據(jù)試劑盒說明進行DNA變性、雜交、連接、PCR擴增;擴增產(chǎn)物在ABI 3130中進行毛細管電泳;應用 Genemapper4.0軟件和 Coffalyser軟件進行分析。
1.2.3 外顯組測序 將先證者全血使用全基因組DNA提取盒(天根DP349,北京)按照產(chǎn)品說明提取基因組DNA,取1 μg DNA 經(jīng)Cavoris儀(美 國 Covaris公 司 )將其打斷至200 bp左右,DNA片段在Klenow Fragment、T4 聚合酶和T4多核苷酸激酶的作用下末端補平修復,其3’端加上A,連接接頭,形成標準的Solexa測序文庫,文庫擴增,與探針雜交,使用鏈霉素磁珠與雜交樣本孵育后洗脫,洗脫產(chǎn)物擴增,經(jīng)Illumina Hiseq2500平臺標準化上機測序。
1.2.4 生物信息學分析 經(jīng)Illumina官方basecall分析軟件BclToFastq得到原始數(shù)據(jù),去除低質(zhì)量的數(shù)據(jù)后,利用BWA(Burrows wheeler aligner)將讀序與人類基因組參考序列(UCSC,Hg19)比對,再分別使用經(jīng)分析軟件 samtools和pindel進行SNP和Indel的過濾篩選,獲得高質(zhì)量可靠的突變。
1.2.5 Sanger法驗證 按照常規(guī)的Sanger法對例9 和例10患者的2個點突變進行測序驗證。擴增引物如下,例9樣本擴增引物為:DD18003793_NNmDeF:TGAGTAGCATATTCCCGTGTCCA,R:CAGAGAACACTCCCCATATCCC;片段長度778bp;例10樣本擴增引物基因DMD-Ex6-seq-F:AATCAGAATAGACTCCTAGCCTT,DMD-Ex6-seq-R:ACTATAACCACTTTCACGCTCC。片段長度599 bp。二者退火溫度均為60度。
2 結(jié) 果
2.1 MLPA分析結(jié)果 應用 MLPA 技術對10例患者血液進行檢測,其中8例檢測到外顯子缺失突變,具體如下:例1外顯子46~52缺失,例2外顯子 45~46、49缺失,例3外顯子 45~47缺失,例4外顯子 44~55缺失,例5外顯子45~47缺失,例6外顯子17~44缺失,例7 外顯子17~44缺失,例8外顯子17~44缺失(見表1),本組中未發(fā)現(xiàn)外顯子重復突變病例。
2.2 外顯組測序 應用外顯組測序結(jié)果及生物信息學分析高通量測序結(jié)果顯示,例9為非編碼區(qū)突變c. 6832-26(IVS49) G>A,例10為點突變c. 116(exon6) G>T(見表2),例9和10同時發(fā)現(xiàn)存在其他基因變異,因與患者臨床表型無相關性,所以這些基因變異暫不考慮與DMD/BMD的致病性有關。
2.3 突變驗證采用Sanger法 對例9~10相應的突變位點進行測序驗證。Sanger法測序與外顯組測序結(jié)果一致 (見圖1) 。

表1 8例BMD/DMD患者基因缺失結(jié)果

表2 2例外顯組及Sanger測序結(jié)果
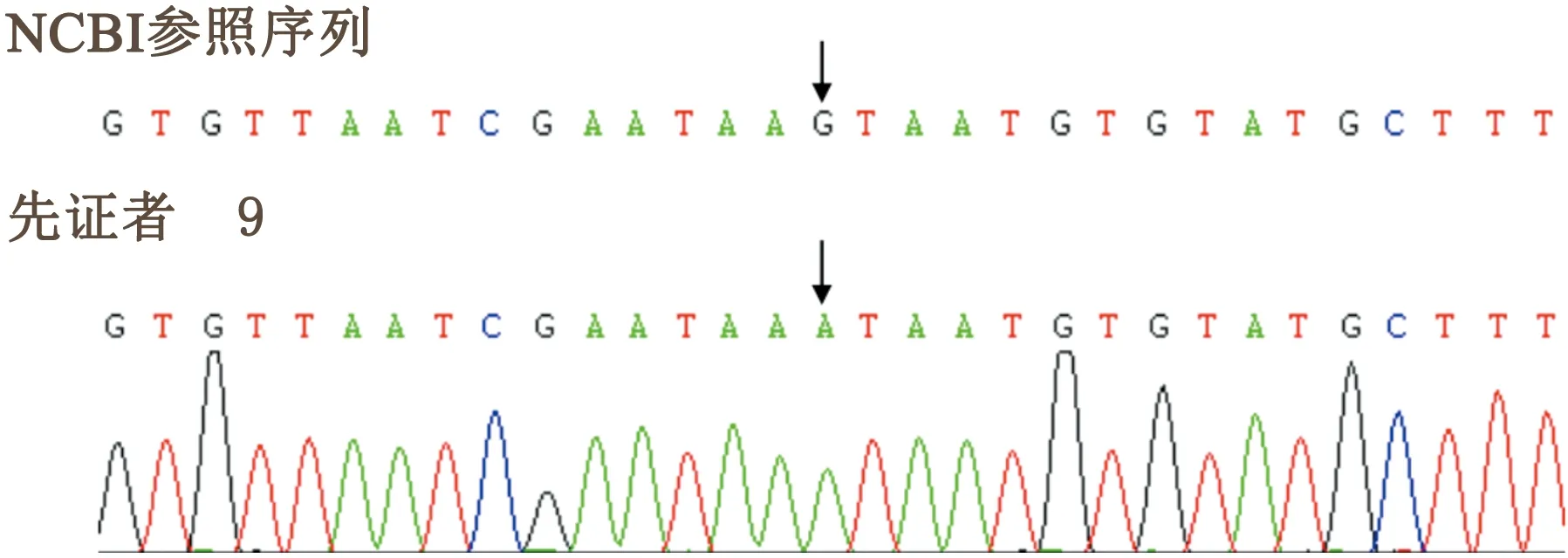
圖1
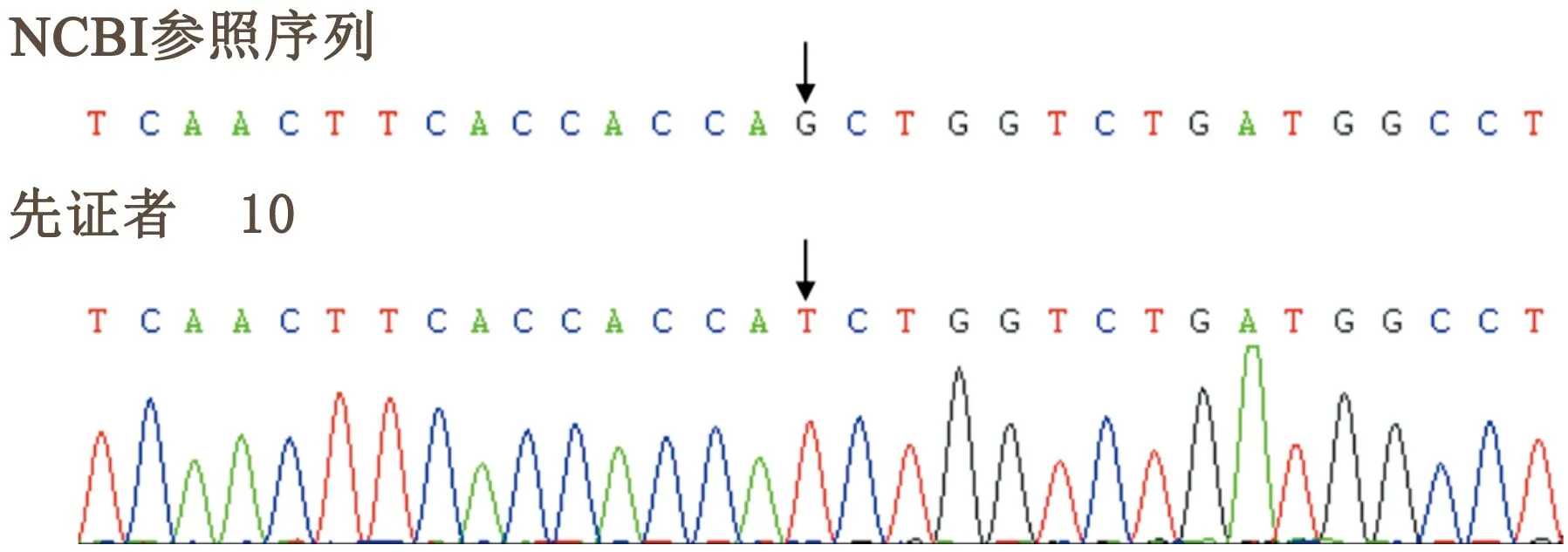
圖2
3 討 論
DMD基因是人體最大的基因之一,位于Xp21.2,全長2400 kb,含79個外顯子,78個內(nèi)含子[6,13]。DMD基因最常見的突變類型為外顯子缺失,約占55%~65%,外顯子重復突變?yōu)?%~10%,其余為點突變或微缺失、插入及剪切位點突變等。致病點突變一般為無義突變或移碼突變[14]。
在本研究中,10例患者有8例外顯子缺失突變,其中缺失外顯子集中于第 44~55外顯子 (見表1),該熱點區(qū)域與多個文獻中報道一致[10,15]。1988年報道指出DMD/BMD多為肌肉組織中抗肌萎縮蛋白(dystrophin)嚴重不足或缺如,而BMD患者則為抗肌萎縮蛋白部分缺乏或分子量異常,因此,BMD較DMD病情較輕[16]。在本研究中,結(jié)合臨床表型,10例患者中有7例診斷DMD,3例診斷BMD。
在MLPA結(jié)果為陰性的2例患者中,我們應用外顯組測序發(fā)現(xiàn)了2種新發(fā)點突變,其中例9為非編碼區(qū)突變c. 6832-26(IVS49) G>A,其致病性尚不明確,例10為點突變c. 116(exon6) G>T,ACMG等級為“致病”。該兩點突變尚未見報道。但其例9,男,24歲,雙下肢無力15 y,蹲下起立困難,尚可獨立行走,鴨步,Gower陽性;例10,男,63歲,雙下肢無力20 y余,近端肢體無力為主,鴨步,其表型符合BMD。此外,例9和10同時發(fā)現(xiàn)其他基因變異,因與患者臨床表型不符,所以這些基因變異暫不考慮與BMD發(fā)病有關。
MLPA和Sanger測序分別被認為是檢測基因外顯子變異和點突變的“金標準”,然而需要事先確定測序的靶向位點以及檢測范圍有限,限制了其在臨床上的廣泛應用。目前,高通量、快速并且已廣泛應用于臨床的NGS已能夠用于DMD的點突變檢測[10,11]。在臨床上,診斷DMD/BMD時,臨床表現(xiàn)為肢體無力,體征有腓腸肌肥大、Gower征陽性,化驗肌酶升高,尤其是肌酸激酶高出數(shù)十倍,肌肉活檢可見肌纖維大小不等,可見肌纖維壞死,抗肌萎縮蛋白免疫組化染色在DMD/BMD患者的肌膜表達完全缺失或部分缺失。基于上述情況,我們可以進行基因檢測:首先選擇MLPA對患者DMD基因進行缺失/重復突變篩查;對于MLPA檢測為陰性者,應用外顯組測序,發(fā)現(xiàn)點突變和微小突變,并Sanger測序進行驗證。以此,規(guī)范DMD/BMD基因診斷流程。為患者提供遺傳咨詢,更為患者日后選擇基因治療做好準備。
猜你喜歡
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
電子制作(2018年18期)2018-11-14 01:48:24
山東工業(yè)技術(2016年15期)2016-12-01 05:31:22
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
中國中醫(yī)藥現(xiàn)代遠程教育(2014年11期)2014-08-08 13:23:44
