舒肝和胃丸質量標準研究
2019-12-03 05:51:38單婷婷張貴英
藥學實踐雜志 2019年6期
胡 丹,尹 明,曹 紅,單婷婷,張貴英
(1.聯勤保障部隊藥品儀器監督檢驗總站 北京100166;2.解放軍總醫院第二醫學中心急診科 北京 100853)
舒肝和胃丸是由醋香附、白芍等十三味藥材組成的中藥復方制劑,用于肝胃不和、兩肋脹滿等。該品種是《中國藥典》2015年版一部品種,原檢驗項目設置較全面,但通過實驗,將薄層部分進行優化、整合,增加了木香、佛手和陳皮藥材的TLC鑒別。文獻[1-3]只有芍藥苷、橙皮苷單純的指標性含量測定的方法,不能全面反映藥品的質量。文獻[4-9]表明白芍總苷( TGP) 是白芍的有效部位,含有芍藥苷、芍藥內酯苷(albiflorin)等一系列單萜糖苷類化合物。已經證明白芍總苷的藥理作用多樣,其中芍藥內酯苷為白芍的特征性成分,在補血、抗抑郁、糖尿病潛在治療方面優于芍藥苷;1,2,3,4,6-O-五沒食子酰葡萄糖有良好的抗腫瘤效果。去甲異波爾定屬于異喹啉生物堿,是中藥材烏藥的主要藥效之一,具有較強的藥理活性,針對關節炎也有很好的療效;橙皮苷是一種天然的二氫黃酮苷,屬黃烷酮類化合物,是陳皮和佛手的主要活性成分之一且性質穩定,在該方劑中含量較高。考慮到上述幾種成分較強的藥理學作用和含量較高等原因,測定單一成分難以反映藥材整體質量,故嘗試建立了HPLC法測定同一供試品溶液中去甲異波爾定、芍藥內酯苷、芍藥苷、1,2,3,4,6-O-五沒食子酰葡萄糖與橙皮苷5種成分的方法。
1 儀器與試藥
1.1 儀器
高效液相色譜儀Agilent 1200(美國,含DAD檢測器);電子天平XS-205DU(METTLER TOLEDO);超聲波清洗器KQ-300E(昆山市超聲儀器有限公司)。
1.2 材料
對照品:去甲異波爾定(批號:111825-201402,95.7%),芍藥苷(批號:110752-200912,98.7%),橙皮苷(批號:110721-201316,95.1%),均購于中國藥品生物制品檢定研究院。芍藥內酯苷(批號:76610010,98.72%)1,2,3,4,6-O-五沒食子酰葡萄糖(批號:76620010,99.39%)均購于上海安譜試驗科技股份有限公司
對照藥材: 木香(批號:120921-201309);陳皮(批號:120921-201309);白芍(批號:120905-201109);佛手(批號:120933-201405);均購于中國藥品生物制品檢定研究院。
試劑:乙腈、甲醇為色譜純,水為超純水,其他試劑均為分析純。
藥品:舒肝和胃丸水蜜丸(A企業,批號16032993);(B企業,批號160303);(C企業,批號20160502);均為9g/丸。
2 方法與結果
2.1 薄層鑒別
2.1.1木香、佛手
取本品水蜜丸5 g,研碎,加入石油醚(30℃~60℃)約60 ml,靜置30 min,超聲30 min,濾過,濾液揮干,殘渣加乙酸乙酯1 ml使溶解,作為供試品溶液。取木香、佛手對照藥材各1 g,加石油醚(30℃~60℃)10 ml,分別按上述方法制成對照藥材溶液。另按處方量分別制備缺木香、佛手的陰性對照品溶液。按薄層色譜法(通則0502)試驗,分別吸取上述4種溶液各5 μl,點于同一硅膠G薄層板上,以乙酸乙酯-甲苯(1∶15)為展開劑,展開,取出,晾干。在日光下檢視,供試品色譜中,在與對照藥材色譜相應的位置,顯相同顏色的斑點(木香)。在紫外(365 nm)下檢視,供試品色譜中,在與對照藥材色譜相應的位置,顯相同顏色的斑點(佛手)。陰性無干擾,結果見圖1、圖2。
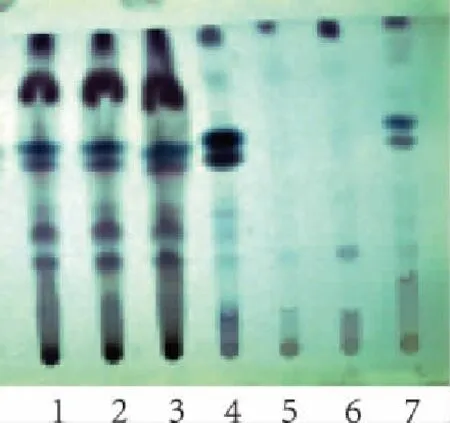
圖1 木香薄層色譜圖 (青島海洋板,溫度:23℃ 相對濕度:35%) 1~3(S1~S3).供試品;4.木香藥材;5.缺木香陰性對照; 6.佛手藥材;7.缺佛手陰性對照
取本品水蜜丸7g,研碎。加甲醇50ml,回流1 h,濾過,取濾液1ml作為供試品溶液,其余備用。另取橙皮苷對照品,加甲醇制成飽和溶液,作為對照品溶液。另按處方量分別制備缺陳皮和佛手的陰性對照品溶液。按照薄層色譜法(通則0502),吸取上述3種溶液各5μl,分別點于同一硅膠G薄層板上,以三氯甲烷-甲醇-水(28∶10∶1)為展開劑,展開,取出,晾干,噴以1%三氯化鋁溶液,置紫外光燈(365 nm)下檢視。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點。陰性無干擾,結果見圖3。
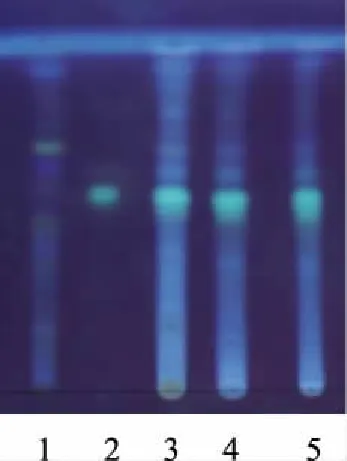
圖3 橙皮苷薄層色譜圖 (青島海洋板,溫度:23℃ 相對濕度:35%)1.陰性對照品;2.對照品;3~5(S1~S3).供試品
2.2 含量測定
2.2.1色譜條件
色譜柱: CAPCELL PAK C18(5 μm ,4.6 mm×250 mm );流動相:0.2%磷酸 (A)-乙腈(B) (v/v);采用梯度洗脫,程序如下:0~30 min,10% B;31~45 min,11%~15% B;46~59 min,16%~19% B;60~74 min,20%~21% B,流速:1.0 ml/min;檢測波長:283 nm;柱溫:35 ℃,進樣體積:10 μl;
2.2.2對照品溶液的制備
取去甲異波爾定、芍藥內酯苷、芍藥苷、1,2,3,4,6-O-五沒食子酰葡萄糖、橙皮苷對照品適量,精密稱定,加稀乙醇制成每1 ml含去甲異波爾定3 μg、芍藥內酯苷10 μg、芍藥苷15 μg、1,2,3,4,6-O-五沒食子酰葡萄糖4 μg、橙皮苷10 μg的混合溶液,即得。
2.2.3供試品溶液的制備
①由經治醫生檢查,簽訂手術同意書,做好手術風險評估。②手術前應該預防感冒、腹瀉等問題。保持大小便通暢。咳嗽、咳痰患者不宜手術。③為確保無菌,避免感染,手術中病人口鼻須遮蓋手術巾,部分患者會有胸悶等不適感,可用毛巾遮蓋口鼻30分鐘進行適當練習,每日2次。
取本品水蜜丸研細,取約0.55g,精密稱定,置具塞錐形瓶中,精密加入稀乙醇25 ml,密塞,稱定重量,超聲處理30 min,放冷,再稱定重量,用稀乙醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.2.4專屬性試驗
按處方工藝,分別制備缺烏藥、佛手、陳皮、白芍的陰性對照樣品。按供試品溶液的制備方法制成各自陰性對照溶液。分別吸取對照品混合溶液、供試品溶液、5種陰性對照溶液各10 μl,注入液相色譜儀,按“2.2.1”項下的色譜條件,進樣測定,記錄色譜圖(見圖4),樣品中去甲異波爾定、芍藥內酯苷、芍藥苷、1,2,3,4,6-O-五沒食子酰葡萄糖、橙皮苷峰與其他組分色譜峰能達到基線分離,理論板數按芍藥苷峰計算不得低于3 000,陰性對照液中色譜峰測定無干擾。
2.2.5線性關系考察
取“2.2.2”項下混合對照品溶液,分別進樣1、5、10、15、20μl注入液相色譜儀,按“2.2.1”項色譜條件測定峰面積,以進樣量(X,μg)為橫坐標,峰面積為縱坐標(Y),繪制標準曲線,計算回歸方程。去甲異波爾定、芍藥內酯苷、芍藥苷、1,2,3,4,6-O-五沒食子酰葡萄糖、橙皮苷的回歸方程,呈良好的線性關系,結果見表1。
2.2.6重復性試驗
取同一批號的舒肝和胃丸(批號16032993)的樣品6份,照“2.2.3”項下方法制備供試品溶液,測定其含量,結果顯示,去甲異波爾定平均含量為0.11 mg/g,RSD為1.83%;芍藥內酯苷平均含量為0.21mg/g,RSD為1.06%;芍藥苷平均含量為0.63mg/g, RSD為1.65%;1,2,3,4,6-O-五沒食子酰葡萄糖平均含量為0.14mg/g,RSD為1.33%;橙皮苷平均含量為1.01mg/g,RSD為1.31%。表明方法的重復性較好。
2.2.7穩定性試驗
取同一批號的舒肝和胃丸(批號16032993)的樣品,照“2.2.3”項下方法配制供試品溶液,精密吸取10 μl,分別在0、4、10、16、22、30 h內進樣,記錄峰面積,結果去甲異波爾定、芍藥內酯苷、芍藥苷、1,2,3,4,6-O-五沒食子酰葡萄糖、橙皮苷峰面積的 RSD分別為1.98%、1.25%、1.44%、1.35%、1.44%。結果表明,舒肝和胃丸的供試品溶液在30 h內基本穩定,表明該方法有良好的穩定性。
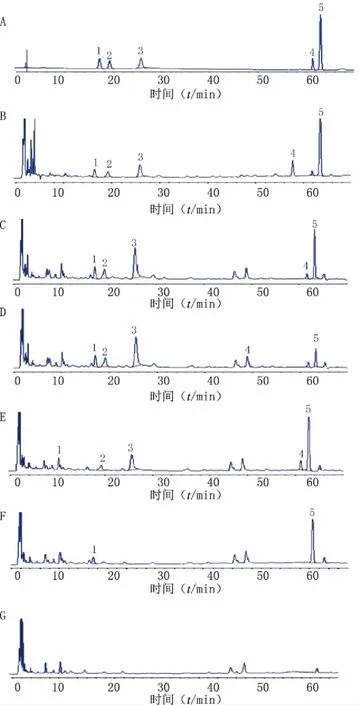
圖4 舒肝和胃丸專屬性HPLC圖譜 A.對照品溶液;B.供試品溶液;C.缺佛手陰性對照溶液; D.缺陳皮陰性對照溶液;E.缺烏藥陰性對照溶液; F.缺白芍陰性對照溶液;G.缺佛手、陳皮、烏藥、白芍的陰性對照溶液 1.去甲異波爾定(烏藥);2.芍藥內酯苷(白芍);3.芍藥苷(白芍); 4.1,2,3,4,6-O-五沒食子酰葡萄糖(白芍);5.橙皮苷(佛手、陳皮)
2.2.8加樣回收率試驗
取同一批號的舒肝和胃丸(批號16032993)的樣品0.275 g,精密稱定,精密加入去甲異波爾定(0.001 28 mg/ml)、芍藥內酯苷(0.002 4 mg/ml)、芍藥苷(0.007 2 mg/ml)、1,2,3,4,6-O-五沒食子酰葡萄糖(0.001 64 mg/ml)、橙皮苷(0.016 mg/ml)混合對照品溶液25 ml,按上述方法制備供試品溶液,按“2.2.1”項下色譜條件,測定其含量,分別計算回收率,結果見表2。
2.2.9樣品測定
按上述方法,制備供試品溶液,依相同色譜條件,以外標法計算,測定3個企業的水蜜丸樣品的含量,結果見表3。

表1 5個對照品的回歸方程、相關系數及線性范圍
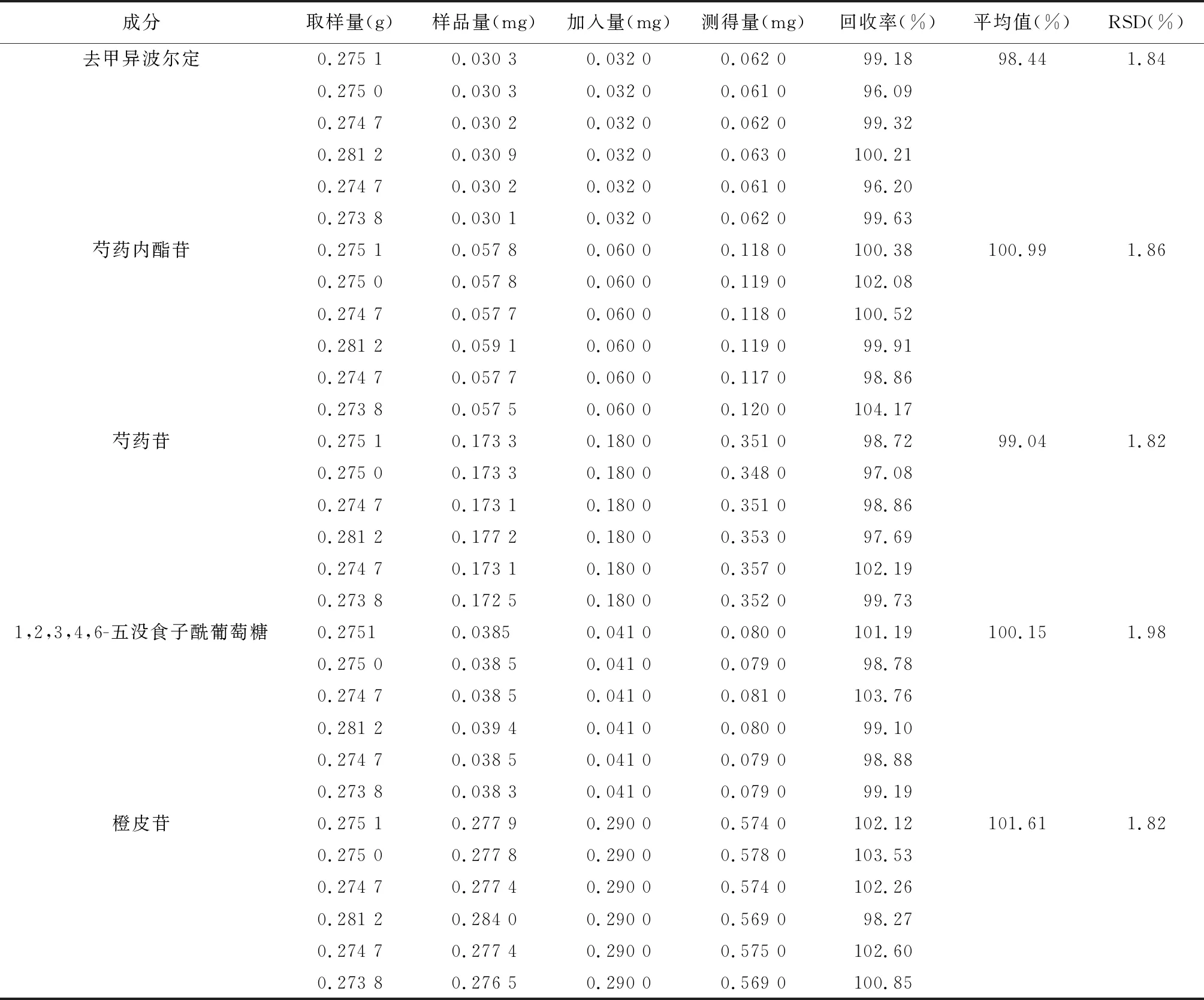
表2 舒肝和胃丸加樣回收率試驗結果(n=6)
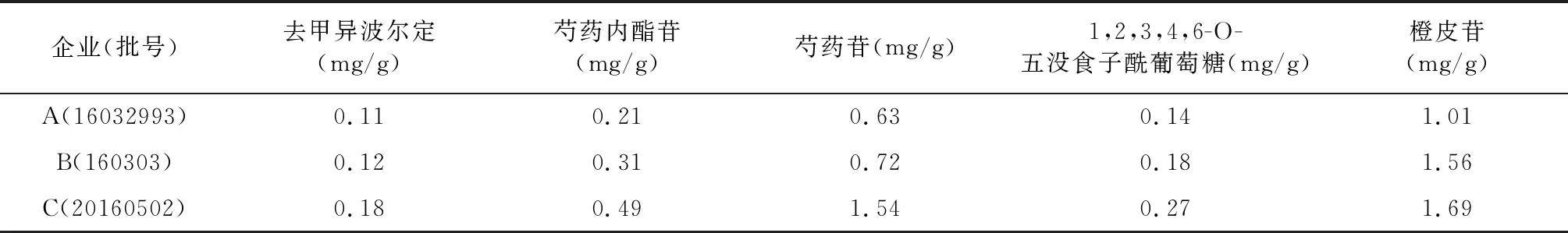
表3 舒肝和胃丸含量測定結果(n=3)
3 討論
3.1 薄層色譜
參照藥典[1],修訂了原方法中佛手的薄層鑒別方法,在此基礎上又同時鑒別木香的TLC法,效果較好,故采用同一供試品溶液分別在日光(鑒別木香)和紫外光(鑒別佛手)下鑒別兩種藥材。其次,陳皮當中含有橙皮苷成分,且佛手藥材中也含有少量的橙皮苷,故同時建立兩者的橙皮苷的TLC法。
3.2 檢測波長的選擇
參考文獻[4-6]5種成分當中有3種成分(芍藥內酯苷、芍藥苷、1,2,3,4,6-O-五沒食子酰葡萄糖)均為白芍藥材的成分,其余2種成分(去甲異波爾定、橙皮苷),經紫外吸收光譜掃描發現最大吸收均為280 nm,考慮到兩者的響應值較高,且5種成分在 230 nm處吸收值相對較好,綜合考慮,最終確定230 nm作為同時測定5種成分的檢測波長。
3.3 提取方法及溶劑選擇
按照中國藥典[1]中測定芍藥苷含量的方法,以芍藥苷峰面積為指標考察回流提取法和超聲提取法對芍藥苷的提取效率的影響,結果顯示,回流與超聲無明顯差異,且回流后樣品黏性大、雜質多,較難過濾,容易損壞色譜柱,最終選擇超聲提取法。分別采用不同濃度的甲醇、乙醇為溶劑提取樣品,最終選取稀乙醇作為供試品溶液的提取溶劑條件。考察了供試品于15、30、45和60 min不同超聲時間的提取結果。結果顯示,30 min后芍藥苷的相對峰面積不再增加,最終選擇超聲30 min作為提取方法。
3.4 流動相選擇
中國藥典[1]采用的緩沖鹽作為流動相,對儀器損傷較大,嘗試用乙腈和0.2%磷酸水、甲醇和0.2%磷酸水、乙腈和水3種試驗方法,結果顯示,乙腈和0.2%磷酸水溶液的梯度洗脫的流動相最為理想,實現了5種成分與相鄰各峰達到有效分離,最終采用該方法,實現節約成本,達到簡便、準確的效果。
綜上所述,本研究修訂了原薄層方法,并建立了“一板多測”的TLC方法和“同一供試品溶液測定多種成分”的HPLC法,具有簡便、穩定可靠、準確等明顯優勢,中藥多組分、多靶點作用機制為生產企業和檢驗部門更加全面的檢測產品質量提供實驗依據。因此作為提高后的質量標準,可以全面控制該品種的質量。
