敲除PaLoc毒力島的無毒性艱難梭菌菌株的構建*
2019-11-06 06:23:12饒鳳琴程玉梅吳昌學王義崔古貞齊曉嵐洪偉
貴州醫科大學學報 2019年10期
饒鳳琴, 程玉梅, 吳昌學, 王義, 崔古貞, 齊曉嵐**, 洪偉**
(1.貴州醫科大學 地方病與少數民族疾病教育部重點實驗室&貴州省分子生物學重點實驗室, 貴州 貴陽 550004; 2.貴州醫科大學附院 綜合ICU, 貴州 貴陽 550004; 3.美國奧本大學 系統生物學系, 美國 奧本 阿拉巴馬 36849; 4.貴州醫科大學 基礎醫學院, 貴州 貴陽 550025)
艱難梭菌(Clostridioidesdifficile)是一種革蘭陽性、產芽孢、專性厭氧棒狀桿菌,在1935年首次由Hall和O`Toole[1]從新生兒糞便樣品中分離得到。艱難梭菌感染(Clostridioidesdifficileinfection, CDI)的主要癥狀有腹瀉、腹痛、發燒、中毒性巨結腸及腸道穿孔等。2002年C.difficile強毒株的出現(核糖體BI/NAP1/027型,同時產生毒素A、毒素B和二元毒素CDT),導致CDI的致病率與致死率顯著提高[2-4],成為醫院獲得性感染中的首要疾病。大約有4%~10%的患者在住院前就攜帶了產毒素艱難梭菌菌株,艱難梭菌表達的毒素A(Toxin A,腸毒素)和毒素B(Toxin B,細胞毒素)是艱難梭菌的主要毒力因子。PaLoc毒力基因島(pathogenicity locus,PaLoc)編碼了2個毒力基因tcdA及tcdB和3個調控基因(tcdR、tcdE及tcdC),3個調控基因共同調控tcdA及tcdB基因的表達。目前發現的毒力艱難梭菌菌株都表達tcdB基因,tcdA基因在某些毒株中選擇性表達。串聯規律間隔短回文序列(cluster regularly interspaced short palindromic repeats, CRISPR)和CRISPR相關蛋白(CRISPR-associated protein, Cas)組成的CRISPR-Cas9系統是一種RNA介導的細菌和古菌免疫系統[5]。在最近幾年,來源于Streptococcuspyogenes的CRISRPR-Cas9元件被廣泛應用于原核生物和真核生物的精確基因編輯[5-19]。與CRISPR-CAS9系統相似,CRISPR-Cpf1系統是基于Cpf1核酸內切酶的免疫系統,該系統最初在FrancisellanocididaU112菌株中被發現[17],后經改造后成為高效的艱難梭菌基因編輯工具[20-21]。本研究使用CRISPR-Cpf1系統成功敲除了艱難梭菌菌株PaLoc毒力島(包含tcdR、tcdB、tcdE、tcdA及tcdC基因),成功構建了去除毒素表達能力的C.difficile菌株,報告如下。
1 材料與方法
1.1 主要菌株、儀器及試劑
菌株NEB express大腸桿菌購于New England BioLabs公司,艱難梭菌 630菌株從美國典型菌種保藏中心(ATCC)購買。儀器及試劑為基因導入儀(Bio-Rad,美國)、厭氧產氣袋(MGC AnaeroPack,日本)、厭氧培養盒(MGC Anaero Pack,日本)、限制性核酸內切酶BtgZI(New England Labs)、NEBuilder?HiFi DNA Assembly Master Mix試劑盒(New England Labs)、腦心浸出液(brain heart infusion, BHI ,Solarbio)、酵母粉(Oxoid,英國)、瓊脂粉(Solarbio)。
1.2 方法
1.2.1菌株培養條件 艱難梭菌放于厭氧培養盒中,并放入厭氧產氣袋,厭氧培養盒置于恒溫培養箱內,37 ℃靜置培養(液體培養8~12 h,固體平板培養18~24 h)。大腸桿菌菌株培養于LB培養基中,必要時添加氯霉素(6 mg/L)及卡那霉素(50 mg/L)。C.difficile培養于補充了5 g /L酵母提取物和1 g/L半胱氨酸腦心浸出液培養基中,37 ℃厭氧培養。適當時候補充以下抗生素/誘導物:甲砜霉素(15 mg/L)、D環絲氨酸(250 mg/L)、頭孢西丁(8 mg/L)及乳糖(40 mmol/L)。
1.2.2質粒構建 本研究中使用的所有質粒及引物見表1。分別用引物擴增PaLoc毒力島上下游同源臂和sRNAP::crRNA,電泳檢測后,膠回收目的條帶。BtgZI酶切載體pWH34,使其線性化。用NEBuilder?HiFi DNA Assembly Master Mix試劑盒將PaLoc毒力島上下游同源臂、sRNAP::crRNA及線性化pWH34載體重組連接,產物電轉化至大腸桿菌,菌落PCR檢測陽性克隆,并計算陽性率(陽性轉化子數與挑取的轉化子數的比值)。將陽性轉化子擴大培養,收取轉化子提取質粒,即為所構建的目的載體。
1.2.3接合轉化艱難梭菌及突變體的篩選 取1 mL含有pWH34質粒的大腸桿菌CA434,6 000 r/min離心3 min,然后用無菌LB液體培養基洗滌2次。細胞沉淀轉移至厭氧箱與200 μL新鮮的艱難梭菌混合。混勻后點至無抗性BHIS瓊脂平板,置于37 ℃厭氧培養10 h,培養平板加入0.5 mL BHIS液體培養基,刮下混合菌液,取100 μL涂布至含有15 mg/L甲砜霉素、250 mg/L D環絲氨酸及8 mg/L頭孢西丁的BHIS瓊脂平板;37 ℃厭氧培養36 h,將菌落挑取并接種到含有15 mg/L甲砜霉素的BHIS培養基中,培養12 h,將細胞進行連續稀釋,取100 μL稀釋涂到包含15 mg /L甲砜霉素及40 mmol/L乳糖的BHIS培養基,37 ℃厭氧培養36 h,隨機挑選菌落并利用菌落PCR篩選。基因組編輯效率為突變體的數量與抗性菌落的比值×100%。
1.2.4質粒丟失 將含有質粒的突變體接種到BHIS培養基中進行再培養(5 d內約轉接10次),然后劃線至固體培養基,挑取單菌落點于BHIS及含有15 mg/L甲砜霉素的BHIS固體培養基(平板影印法)。選擇對甲砜霉素敏感的菌落,在BHIS培養基中進行培養。通過PCR進一步證實這些突變體是否為質粒丟失的突變株。
1.2.5突變株的基因型鑒定 為了驗證突變株基因型的正確性,以表1中的6、7為檢測引物,突變株基因組為模板, 進行PCR驗證,反應條件為94 ℃預變性5 min,94 ℃變性20 s、55 ℃退火30 s、68 ℃延伸2 min,30個循環,68 ℃徹底延伸10 min。電泳鑒定后送至上海生工生物工程有限公司進行測序驗證。

表1 質粒、引物序列及來源
2 結果
2.1 PaLoc毒力島敲除質粒的構建
圖1 A所示為構建pWH53質粒所需基因元件的克隆,其中1、2泳道為PaLoc毒力島上下游同源臂(長度分別為1 048 bp,1 036 bp),3泳道為sRNAP::crRNA片段,4泳道為BtgZI酶切后的線性化pWH34質粒。以上4個片段使用NEBuilder?HiFi DNA Assembly Master Mix試劑盒一步重組連接,轉化大腸桿菌NEB Express感受態細胞。轉化子挑取16個克隆PCR檢測,其中10/16為陽性克隆,這些克隆擴增后提取質粒獲得PaLoc打靶質粒pWH53(圖1B)。該質粒包含可在大腸桿菌-艱難梭菌中穿梭復制的復制子、traJ接合轉化元件、甲砜霉素抗性基因(catP)、iLacP::Cpf1和sRNAP::crRNA表達框,質粒大小為13 055 bp(圖1B)。
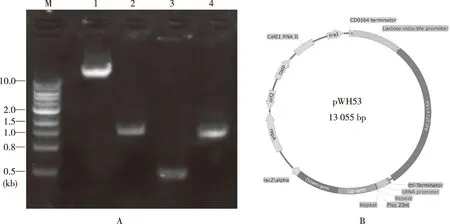
注:A為構建pWH53質粒所需基因元件的克隆,B為克隆擴增后提取質粒獲得PaLoc打靶質粒pWH53。
2.2 pWH53質粒轉化及突變株篩選
包含pWH53質粒的大腸桿菌CA434細胞與艱難梭菌 630細胞進行接合轉化,轉化效率為0.25×103CFU/L。獲得pWH53質粒轉化子后,首先挑取艱難梭菌 630轉化子接入具有甲砜霉素抗性的BHIS培養基中,以維持pWH53質粒在艱難梭菌630菌株中的穩定傳代(圖2A);再將艱難梭菌630轉化子菌液稀釋100~1 000倍后涂布于含有乳糖的BHIS-甲砜霉素培養基中,以誘導Cpf1蛋白的表達(圖2A)。ΔPaLoc基因敲除菌株經在無抗性的BHIS培養基中連續轉接10次后丟失pWH53質粒,獲得穩定的ΔPaLoc突變株。擴增ΔPaLoc突變株與艱難梭菌 630 WT基因組DNA:第1泳道以ΔPaLoc突變株基因組DNA為模板,第2泳道以艱難梭菌 630基因組DNA為模板,第3泳道為以ddH2O為模板的對照。泳道M為DNA質量標準,由于以艱難梭菌 630基因組DNA為模板時擴增片段太長(26 487 bp)因此無擴增條帶(圖2B)。
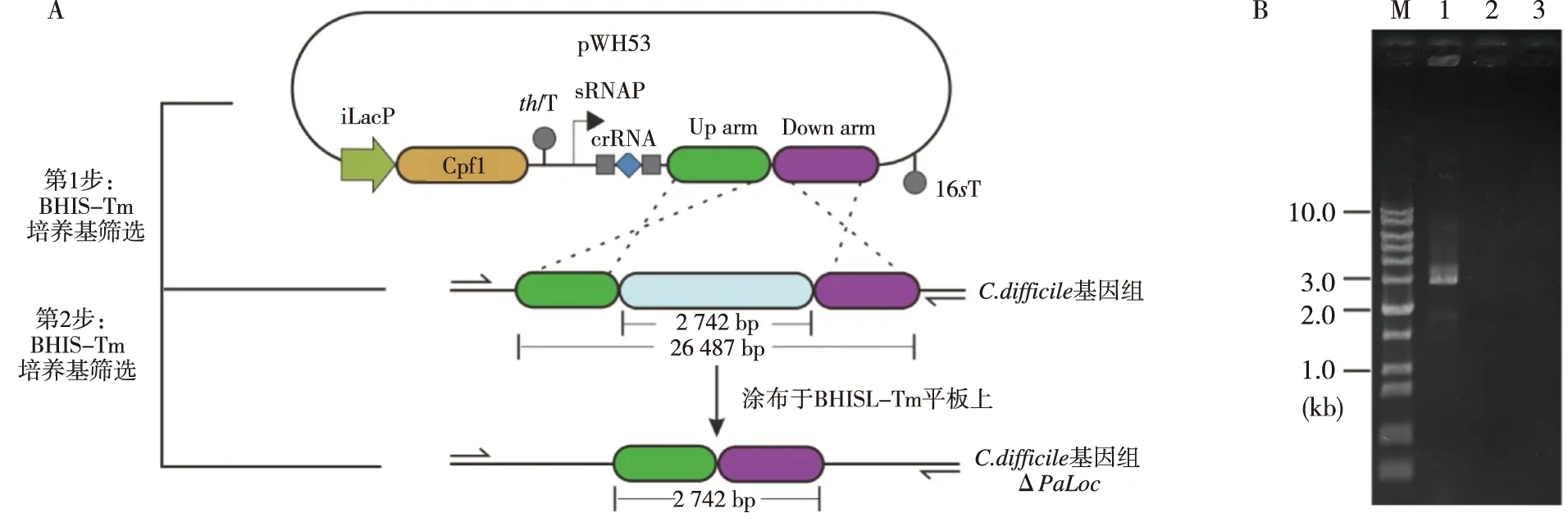
注:A為質粒轉化及突變株篩選示意圖,B為擴增ΔPaLoc突變株及630基因DNA電泳結果,M為DNA mark,1為ΔPaLoc DNA,2為630 DNA,3為對照。
2.3 ΔPaLoc突變株基因型鑒定
將ΔPaLoc突變株獲得擴增片段進行測序,結果如圖4所示。使用Vector NTI軟件比對上游同源臂、下游同源臂與ΔPaLoc突變株上下游同源臂重組位點,發現ΔPaLoc突變株相應位點為上下游同源臂嵌合體序列,說明上下游同源臂之間的PaLoc毒力島被完整敲除(圖3A)。并且相應位置(上下游同源臂的連接點)的測序峰圖文件(圖3B)測序信號清晰,提示ΔPaLoc構建成功。
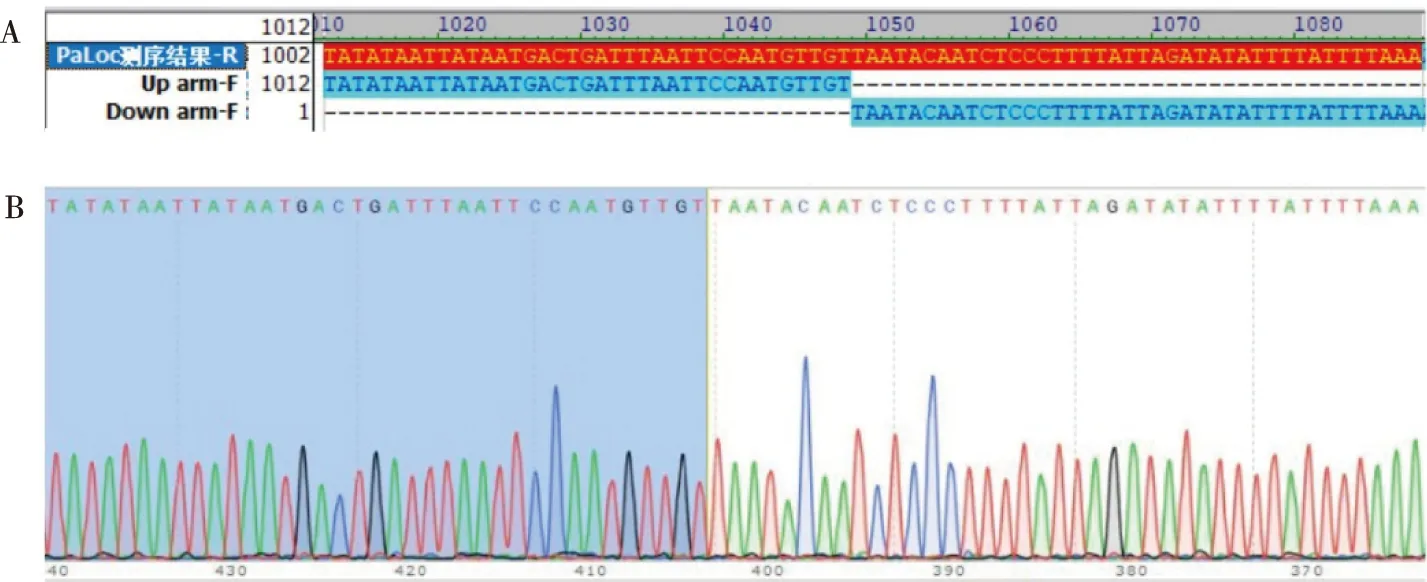
注:A為Vector NTI軟件化對結果,B為測序結果。
3 討論
艱難梭菌是醫院獲得性腹瀉的主要誘因,攝入抗生素的住院患者是CDI的危險人群。艱難梭菌的毒力因子主要由PaLoc毒力基因島編碼,包括tcdR、tcdB、tcdE、tcdA及tcdC5個基因。本研究使用CRISPR-Cpf1技術成功敲除了長達26 487 bp的PaLoc毒力島。相比于CRISPR-Cas9技術[22],CRISPR-Cpf1技術因不需要tracRNA的參與,Cpf1編碼基因更小,因此具有更高的接合轉化效率,另外,CRISPR-Cpf1技術更適合于編輯大基因片段。Hong等[20]報道,使用該技術成功敲除了接近50 kb的基因片段。相比基于反義RNA[23]、Clostron[24-25]、CodA[26]、ACE[27]等技術,CRISPR系統具有更高的基因編輯效率和更操作等優點。
PaLoc毒力島tcdR、tcdB、tcdE、tcdA及tcdC基因中,其中的tcdA及tcdB編碼主要的毒力因子TcdA及TcdB。2種毒素都可以于腸上皮細胞頂端接合,后通過內化作用進入細胞,引起腸細胞骨架變化,導致上皮屏障松動和細胞間連接被破壞。細胞間連接的破壞使得細胞間連接變得松散,TcdA、 TcdB毒素有機會進一步侵入腸上皮細胞。毒素作為遠外源物質會導致吞噬細胞、肥大細胞和各種免疫調節介質,導致炎癥和中性粒細胞的積累,引發炎癥并最終導致CDI癥狀的發生[28]。敲除PaLoc毒素島后突變株不再產生致病毒素,并且該突變株因細胞壁及細胞壁蛋白仍然完整,因此并未丟失免疫原性,因此該ΔPaLoc突變株有望進一步開發為全菌疫苗。
然而,活菌疫苗的構建僅僅敲除ΔPaLoc毒力島可能還不夠。如最近的研究發現,有的強艱難梭菌毒株除了攜帶PaLoc毒力島,表達TcdA、TcdB毒素外,還表達二元毒素肌動蛋白特異性ADP核糖基轉移酶毒素(actin-specific ADP-ribosyltransferase toxin, CDT;如強毒株NAP1/BI/027)。CDT毒素由CdtLoc毒力島編碼,通常位于PaLoc毒力基因島下游,由cdtR、cdtA及cdtB基因組成。cdtA及cdtB基因分別編碼CDTa(酶組分)和CDTb(黏附組分),亦對CDI的發生有貢獻。因此在未來的工作中,課題組將進一步編輯ΔPaLoc突變株敲除CdtLoc毒力島,并進一步完成表型及動物模型試驗。
綜上,本研究結果顯示,在獲得的16株轉化子中,有8株為敲除PaLoc突變株,陽性率為50%;通過測序驗證,獲得的8株敲除PaLoc突變株均在設計位點發生了等位雙交換,成功構建艱難梭菌毒性毒力島敲除PaLoc突變株。
