高效液相色譜法測定雞組織中苯并咪唑類藥物殘留標志物
2019-09-16 08:22:14邵琳智吳映璇
色譜 2019年10期
鄒 游, 邵琳智, 吳映璇
(廣東檢驗檢疫技術中心, 廣東 廣州 510623)
苯并咪唑類(BMZs)化合物是含有兩個氮原子的苯并雜環化合物,具有良好的生物活性[1],同時抗菌、抗病毒、抗寄生蟲等療效明顯[2],被廣泛用于農業、水產養殖和獸醫中。研究表明,BMZs具有致畸與致突變作用,且在體內轉化的代謝產物仍具有毒理作用[3]。因此,中國、美國、歐盟等大多數國家和地區將其作為食品安全重要的監控對象,并制定了最高殘留量(MRL)[4]。
我國最新出臺的《食品安全國家標準動物性食品中獸藥最大殘留限量》(征求意見稿)[5]較之前的農業部235號公告[6]對苯并咪唑類藥物作了較大修改,不僅增加了藥物的種類,而且規范了名稱,其次對殘留標志物也作了修改:非班太爾、芬苯達唑和奧芬達唑的殘留標志物為芬苯達唑、奧芬達唑和奧芬達唑2-氨基砜的總和;奧苯達唑的殘留標志物為奧苯達唑;噻苯達唑的殘留標志物為噻苯達唑和5-羥基噻苯達唑;三氯苯達唑的殘留標志物為三氯苯達唑酮;阿苯達唑的殘留標志物為阿苯達唑-2-氨基砜;氟苯達唑的殘留標志物為氟苯達唑;甲苯達唑的殘留標志物為2-氨基-5-苯甲酰基苯并咪唑和5-羥基甲苯達唑,共涉及9種苯并咪唑類藥物,對應殘留標志物共11種。
苯并咪唑類藥物因其化學結構的關系,一般呈弱堿性,但其中三氯苯達唑的殘留標志物三氯苯達唑酮為中性化合物,若要同時檢測這9種苯并咪唑類藥物的殘留標志物,技術上存在許多困難,尤其是采用高效液相色譜法時,對于樣品的凈化具有更高的要求。然而,不管是已頒布的國家標準[7,8]或行業標準[9],還是已發表的文獻或研究[10-18],都尚未有完全包括這11種殘留標志物的檢測方法公開。本文采用的前處理方式具有獨創性,尤其是凈化技術,解決了三氯苯達唑酮等幾種殘留標志物不保留、回收率低的問題。本方法為雞組織中苯并咪唑類藥物殘留標志物測定標準的制定提供了可靠的參考依據,具有廣闊的應用前景。
1 實驗部分
1.1 儀器、試劑與材料
LC-20AD高效液相色譜儀,配自動進樣器、柱溫箱、紫外檢測器和二極管陣列檢測器(日本島津公司);感量0.000 01 g的分析天平和感量0.01 g的天平(德國Sartorius公司); GM 200型肉類組織搗碎機(德國Retsch公司); MS 3 basic旋渦振蕩器、減壓旋轉蒸發儀(德國IKA公司); SW 30H超聲波水浴(瑞士Sono Swiss公司); 3-30K離心機(德國Sigma公司)
奧芬達唑(oxfendazole, CAS號:53716-50-0)、芬苯達唑(fenbendazole, CAS號:43210-67-9)、奧芬達唑-2-氨基砜(oxfendazole sulphone, CAS號:54029-20-8)、阿苯達唑-2-氨基砜(albendazole-2-aminosulfone, CAS號:80983-34-2)、噻苯達唑(thiabendazole, CAS號:148-79-8)、5-羥基噻苯達唑(5-hydroxythiabendazole, CAS號:948-71-0)、2-氨基-5-苯甲酰基苯并咪唑(mebendazole-amine, CAS號:52329-60-9)、5-羥基甲苯達唑(5-hydroxymebendazole, CAS號:60254-95-7)、氟苯達唑(flubendazole, CAS號:31430-15-6)、奧苯達唑(oxibendazole, CAS號:20559-55-1)和三氯苯達唑酮(ketotriclabendazole, CAS號:1201920-88-8),含量均在97%以上(德國Dr. Ehrenstorfer公司);乙腈、甲醇(色譜純,美國Fisher公司),乙酸(色譜純,德國Merck公司);實驗室用水為超純水(符合GB/T 6682-2008一級水要求);其他未作特殊說明的試劑均為分析純;Oasis MCX固相萃取柱(60 mg/3 mL,美國Waters公司)。
1.2 樣品前處理
1.2.1提取
稱取5 g試樣(準確至0.01 g),置于50 mL離心管中,加入15 mL乙腈溶液,超聲5 min,渦旋振蕩15 min,以4 000 r/min離心5 min,上清液轉移至25 mL容量瓶中;殘渣再加入10 mL乙腈溶液,重復上述過程,合并上清液,并用乙腈定容至刻度,搖勻,備用。
1.2.2凈化
取5 mL上述提取液,置于15 mL塑料離心管中,加入5 mL 0.1 mol/L鹽酸溶液,再加入5 mL正己烷,渦旋振蕩1 min,以10 000 r/min離心5 min,棄去上層正己烷層,下層清液加入5 mL 0.1 mol/L鹽酸溶液混勻,轉入經過預處理的MCX固相萃取柱中,以小于1 mL/min的流速使樣品溶液全部通過小柱,棄去流出液。依次用3 mL 0.1 mol/L鹽酸-甲醇(2∶1, v/v)、4 mL 5%(v/v)氨水溶液淋洗,淋洗液全部通過小柱,棄去淋洗液。最后用4 mL 5%(v/v)氨水甲醇以0.5 mL/min的流速洗脫,收集洗脫液于具刻度的濃縮瓶中。
1.2.3濃縮、定容
將收集的洗脫液于38 ℃水浴下減壓旋轉蒸發至約500 μL,加入0.1 mol/L鹽酸溶液定容至1 mL,渦旋2 min,以10 000 r/min離心5 min,待測定。
1.3 色譜條件
色譜柱:Atlantis T3色譜柱(250 mm×4.6 mm, 5 μm);柱溫:40 ℃;流動相:A為甲醇溶液,B為1%(v/v)乙酸水溶液;流速:1.0 mL/min;進樣量:40 μL;檢測波長:292 nm。洗脫梯度見表1,梯度均為線性變化。
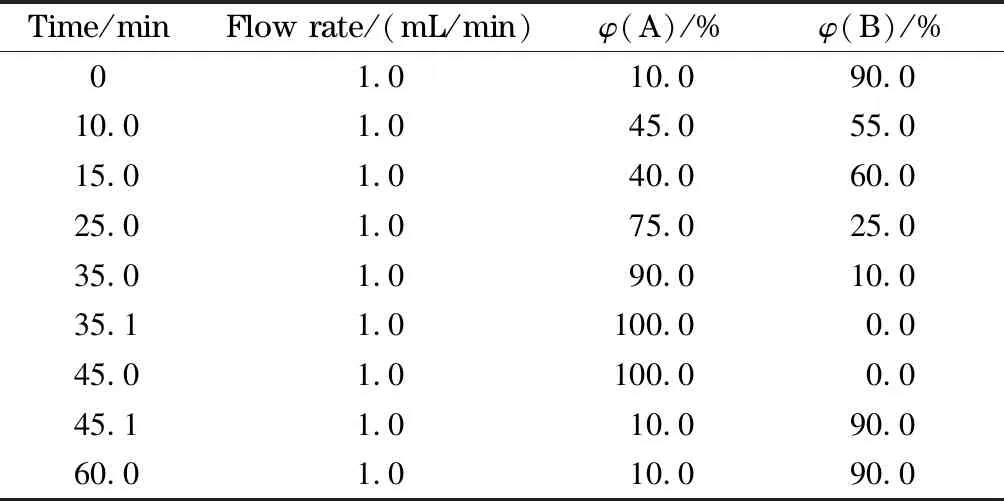
表 1 梯度洗脫程序
A: methyl alcohol; B: 1% (v/v) acetic acid aqueous solution.
2 結果與討論
2.1 色譜條件的優化
苯并咪唑類藥物屬于弱堿性物質,中等極性,現有國家標準和參考文獻多采用C18柱進行檢測[7-9]。針對極性較小的化合物,普通C18柱保留較弱,本方法嘗試選擇Atlantis T3色譜柱進行分離,其鍵合和端基封尾技術的結合,不僅對極性化合物保留出色,且更加穩定耐用,較好地改善了普通C18色譜柱堿性化合物分離不佳、柱壽命較短等狀況。
本方法在選擇流動相時,比較了有機相乙腈和甲醇溶液的分離與保留效果,發現并無太大差異,但使用乙腈作為有機相洗脫時會產生較多雜峰,使用甲醇時則雜峰較少。對于本方法中的11種苯并咪唑類藥物殘留標志物,流動相中不同酸度的水相對其分離和保留的影響較大。本方法嘗試了采用0.5%、1%和2%體積分數的乙酸水溶液,發現采用1%(v/v)乙酸水溶液時,全部待測化合物的分離和保留達到最佳。因此本方法最終采用甲醇-1%(v/v)乙酸水溶液進行梯度洗脫。經過色譜優化后,得到的11種苯并咪唑類殘留標志物標準溶液(100 μg/L)、空白雞肝臟樣品和加標雞肝臟樣品(500 μg/L)的高效液相色譜圖見圖1。
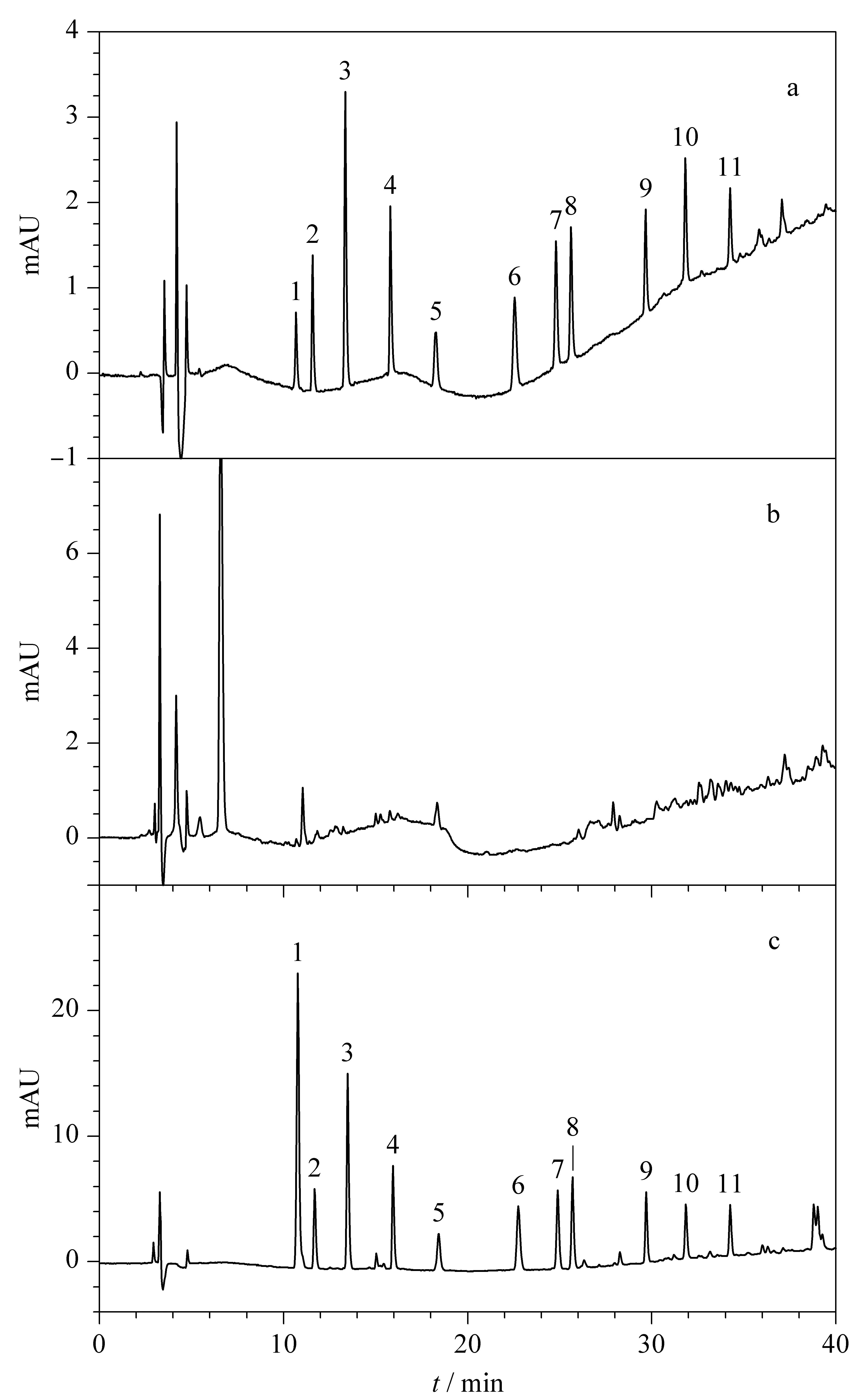
圖 1 (a)11種苯并咪唑類殘留標志物標準溶液 (100 μg/L)、(b)空白雞肝臟樣品和(c)加標雞肝臟樣品(500 μg/L)的色譜圖 Fig. 1 (a) Chromatograms of the 11 benzimidazoles residue marker standard solution (100 μg/L), (b) blank chicken liver sample and (c) spiked chicken liver sample (500 μg/L) 1. albendazole-2-aminosulfone; 2. 5-hydroxythiabendazole; 3. thiabendazole; 4. mebendazole-amine; 5. 5-hydroxymebendazole; 6. oxibendazole; 7. oxfendazole; 8. oxfendazole sulphone; 9. flubendazole; 10. fenbendazole; 11. ketotriclabendazole.
2.2 提取溶劑的選擇
與大多數標準和文獻所采用的提取溶劑不同,本文比較了乙腈、甲醇、80%(v/v)乙腈水溶液、二氯甲烷、乙酸乙酯或堿性乙酸乙酯(含0.15 mL 50%(質量分數)氫氧化鉀水溶液,下同)[7]的提取效果。使用甲醇、80%(v/v)乙腈水溶液或二氯甲烷提取時,芬苯噠唑、奧芬噠唑等目標物質的回收率低于50%;使用乙腈、乙酸乙酯或堿性乙酸乙酯時,所有化合物的提取效率都超過90%。但在本方法的色譜條件下,乙腈提取時干擾峰遠遠少于采用乙酸乙酯或堿性乙酸乙酯時的干擾峰。另外,乙酸乙酯或堿性乙酸乙酯提取液在后一步凈化上柱前必須進行溶劑轉換,常用方法為吹氮濃縮或減壓旋轉蒸發濃縮至干,再用乙腈-0.1 mol/L鹽酸(1∶2, v/v)溶液溶解。然而濃縮過程會引起三氯苯達唑酮的降解;而采用乙腈提取則省去濃縮步驟,加入0.1 mol/L鹽酸溶液后即可上柱,且乙腈提取液對所有化合物的提取效率均可達到95%以上。因此本文選擇乙腈作為提取溶劑。
2.3 凈化方式的優化
本方法比較了C18、HLB和MCX固相萃取小柱的萃取效果,使用C18和HLB小柱,且上樣溶液中乙腈的體積分數大于10%時,奧芬達唑-2-氨基砜和5-羥基噻苯達唑會有所損失,故乙腈提取液必須先進行濃縮,而濃縮又會導致待測化合物降解,且復溶解時有機溶劑含量低,溶解效果差,也會導致回收率降低,所以這兩種固相萃取小柱均不適用。本方法選擇采用MCX小柱,在乙腈體積與水相體積之比不超過三分之一的情況下,所有待測化合物在固相萃取柱上都能全部保留,提取液無需濃縮,可稀釋后直接上樣。
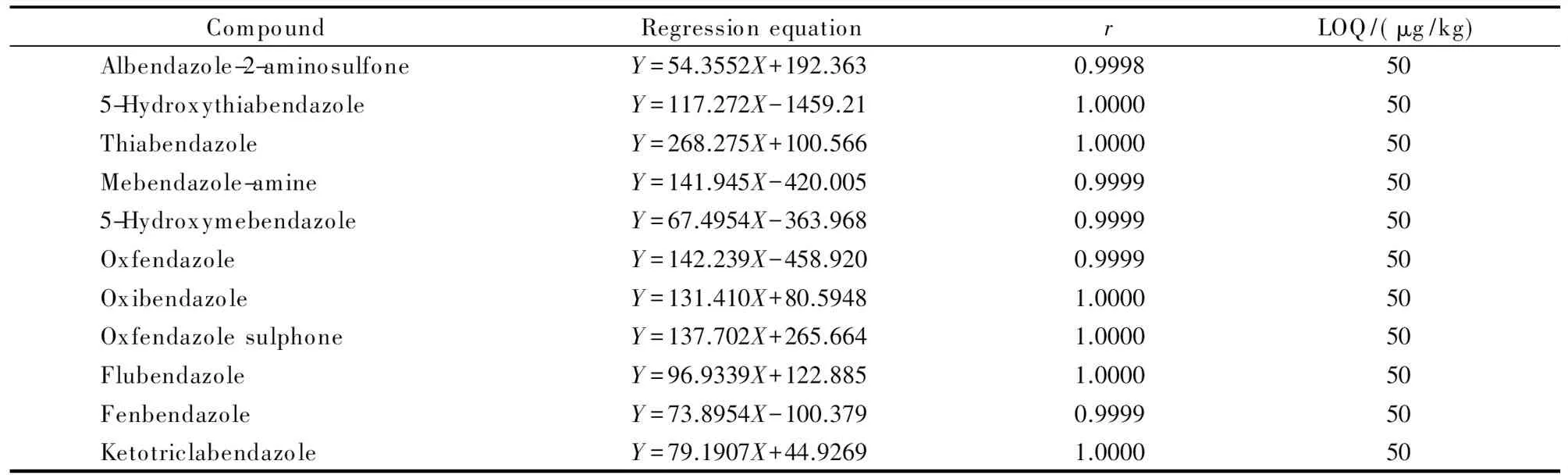
表 2 11種苯并咪唑類藥物殘留標志物的回歸方程、相關系數(r)和定量限
Y: peak area;X: mass concentration, μg/L.
采用MCX固相萃取小柱進行凈化時,11種待測化合物存在兩種不同的保留機理,對于三氯苯達唑酮而言,填料中的N-乙烯基吡咯烷酮-二乙烯苯共聚物起反相吸附的作用;而對于其余10種化合物而言,不僅存在反相吸附作用,填料中共聚物基質上鍵合的磺酸基還起到離子交換的作用。故對于上柱、淋洗和洗脫溶液會有更多限制條件,需要兼顧溶液的酸堿性和其中有機溶劑所占的體積分數。按照GB/T 21324-2007《食用動物肌肉和肝臟中苯并咪唑類藥物殘留量檢測方法》中所用的過柱條件處理加標樣品,三氯苯達唑酮會隨淋洗液流出,導致回收率為零。在本方法的上柱溶液中,乙腈與0.1 mol/L鹽酸溶液的體積比為1∶2,若比例為1∶1,三氯苯達唑酮幾乎不被小柱保留,損失大半,而淋洗使用的0.1 mol/L鹽酸-甲醇溶液中甲醇的體積分數也不宜超過30%;另一淋洗溶液5%(v/v)氨水溶液能有效去除奧芬達唑-2-氨基砜和5-羥基噻苯達唑兩個峰之間的雜質,若采用0.1 mol/L鹽酸溶液則不能去除該雜質;若用氨水乙腈洗脫,氨的體積分數對2-氨基-5-苯甲酰基苯并咪唑的影響較大,采用5%(v/v)氨水乙腈溶液洗脫,回收率約為50%,氨的體積分數升高,非常容易揮發,導致方法的重現性不好。最終,選擇了對各組分的洗脫能力比較穩定且后續濃縮也更為容易的5%(v/v)氨水甲醇作為洗脫液。
2.4 方法學驗證
2.4.1靈敏度
在空白雞肉、雞肝臟和雞腎臟樣品中分別添加一定濃度的苯并咪唑類藥物殘留標志物標準溶液,測定其S/N,以S/N≥10且回收率和相對標準偏差均符合殘留檢測方法要求時的含量為定量限。以峰面積(Y)對其質量濃度(X, μg/L)進行線性回歸,繪制標準工作曲線(見表2)。結果表明,11種苯并咪唑類藥物殘留標志物在25~1 000 μg/L范圍內線性關系良好,定量限能夠滿足相關監管部門的要求。
2.4.2準確度和精密度
準確稱取5.0 g空白試樣,置于50 mL離心管中,加入已知濃度的系列標準工作液,制成低、中和高3個不同水平的雞肉、雞肝和雞腎組織樣品,每個水平做6個平行樣品,按樣品前處理過程處理后分析,計算加標回收率(見表3)。結果表明,11種苯并咪唑類藥物的加標回收率為70.91%~103.21%, RSD為1.48%~10.08%。
2.5 實際樣品測定
采用本方法檢測雞肉、雞肝臟及雞腎臟樣品共150份。結果顯示,1個雞肝臟樣品檢出阿苯達唑-2-氨基砜,含量為78 μg/kg,其余樣品均未檢出相關物質。
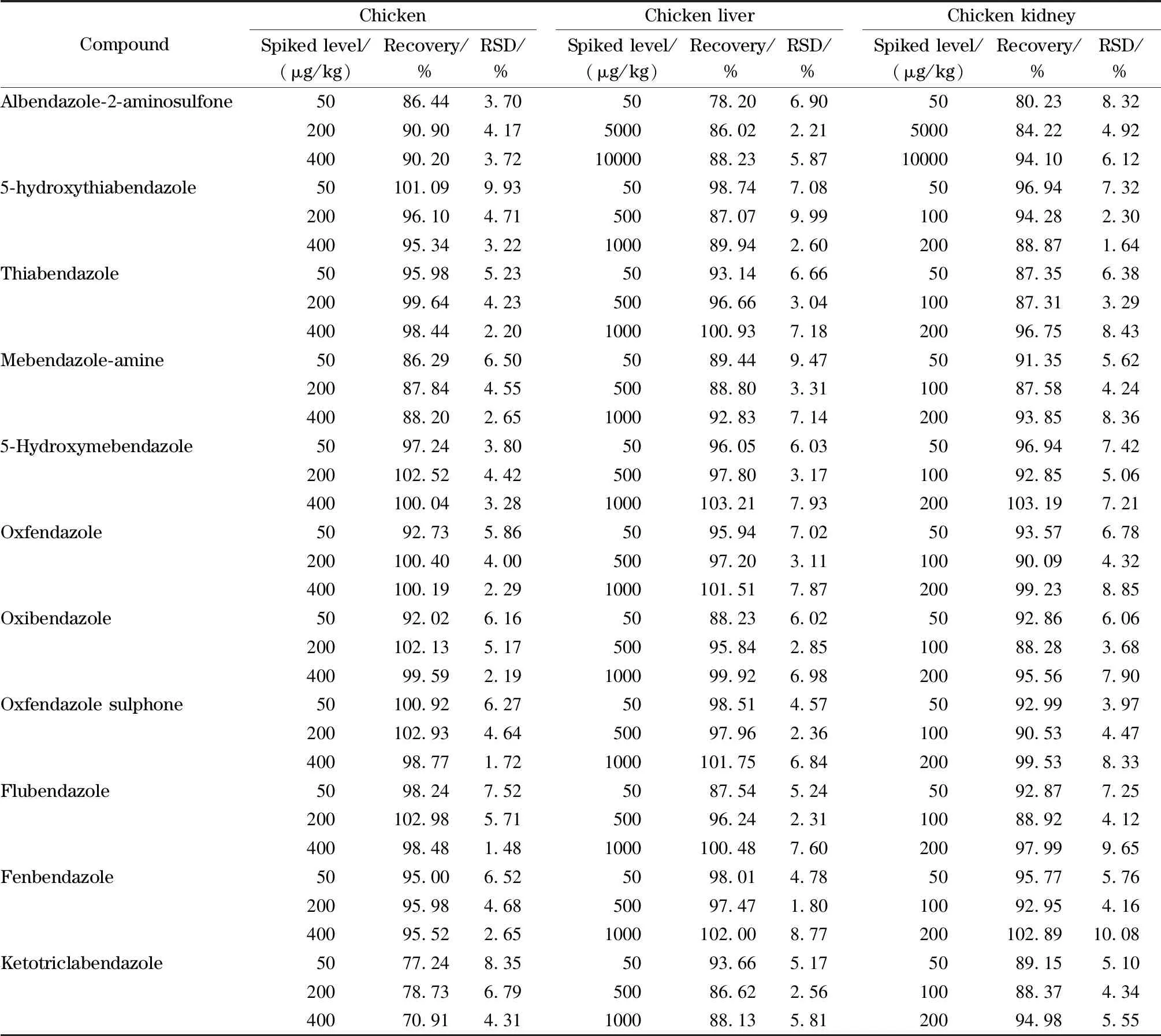
表 3 雞肉、雞肝臟和雞腎臟樣品中11種苯并咪唑類藥物殘留標志物的加標回收率及相對標準偏差(n=6)
3 結論
本文建立了高效液相色譜同時測定雞肉、雞肝臟和雞腎臟中苯并咪唑類藥物殘留標志物的檢測方法。該方法定量科學準確,抗干擾能力強,基質適應范圍廣,可廣泛應用于雞組織中苯并咪唑類藥物殘留的日常監測,能夠滿足我國相關監管部門的最新技術要求,為管理部門提供可靠有效的技術支持。
