QuEChERS-氣相色譜-串聯質譜法測定枸杞中農藥殘留
2019-09-16 08:22:10王芳煥任翠娟郝俊虎
色譜 2019年10期
王芳煥, 任翠娟, 馬 輝, 李 萍, 郝俊虎, 陳 林, 孫 敏
(銀川海關技術中心, 寧夏 銀川 750002)
枸杞是寧夏傳統出口農產品,具有極高的營養價值,不僅含有豐富的微量元素,而且含有大量糖、脂肪、蛋白質、色素和維生素等物質[1-3],也正是因為營養豐富,易受到病蟲害威脅,而有效防治病蟲害,必須使用化學農藥。但是枸杞生產過程中,農藥使用不合理,易造成枸杞產品農藥殘留問題。近幾年,出口日本和美國的枸杞干果,其監控的擬除蟲菊酯類農藥的殘留限量標準分別為0.5和0.01 mg/kg,銷往香港和臺灣的枸杞干果,其監控的擬除蟲菊酯類、有機磷類、殺螨劑、殺蟲劑等農藥殘留項目均不得檢出。然而出口枸杞中檢出農殘被官方通報的情況時有發生,包括啶蟲脒、毒死蜱、三唑酮等25種農殘項目。隨著枸杞貿易地區對農藥殘留監控日益嚴苛,農藥殘留已成為當前影響我國枸杞質量安全、出口創匯的主要因素。為了考察枸杞中農藥的殘留情況,本實驗室收集了代表性的枸杞樣品,采用GC-MS/MS方法,對400種農藥殘留進行了初步篩查,然后結合我國枸杞主要貿易國家或地區監控的農藥殘留項目,選取了有機磷、擬除蟲菊酯、殺螨劑、殺蟲劑等20種農藥,進行方法研究并建立定量方法。
目前農藥多殘留快速檢測是理化分析研究熱點,主要分為樣品前處理技術改進和儀器檢測條件優化。2003年,Anastassiades等[4]在基質固相分散的基礎上開發了QuEChERS方法,其具有靈敏、高效、快速等特點,近年來被廣泛應用。經典的QuEChERS方法雖簡便快捷,但采用吸附劑種類少,無法去除色素、糖類、脂肪等對質譜干擾較大的雜質。本文分別考察了乙二胺-N-丙基硅烷(PSA)、C18和石墨化炭黑(GCB)3種吸附劑對枸杞樣品中雜質的去除效果,最終優化出最佳的吸附劑量。
目前多農藥殘留的檢測方法主要有分光光度計法[5]、酶聯免疫測定法[6]、生物傳感器測定法[7]、氣相色譜法(GC)[8]、液相色譜法[9]、高效液相色譜-質譜聯用法(LC-MS/MS)[10-13]、氣相色譜-質譜聯用法(GC-MS)[14-18]。在實際應用中,GC易受基質干擾,確證不足;GC-MS在檢測分子量低或者沸點較高的農藥時,易受基質成分和柱流失干擾,影響檢測結果。本文采用GC-MS/MS檢測枸杞中20種農藥殘留,不僅消除了枸杞中復雜基質的干擾,而且實現了20種農藥的快速、準確定量。
1 實驗部分
1.1 儀器、試劑與材料
Agilent7000A氣相色譜-質譜/質譜儀(美國安捷倫科技有限公司), TDL-80-2B離心機(上海安亭科學儀器廠), QT-2A迷你漩渦振蕩器(上海琪特分析儀器有限公司), BD-202電子天平(梅特勒(上海)有限公司), CPA225D電子天平(德國賽多利斯公司)。甲胺磷(methamidophos, CAS號:10265-92-6)、甲拌磷(phorate, CAS號:298-02-2)、甲基毒死蜱(chlorpyrifos-methyl, CAS號:5598-13-0)、馬拉硫磷(malathion, CAS號:121-75-5)、毒死蜱(chlorpyrifos, CAS號:2921-88-2)、對硫磷(parathion, CAS號:56-38-2)、三唑酮(triadimefon, CAS號:43121-43-3)、三唑醇(triadimenol, CAS號:55219-65-3)、氟蟲腈(fipronil, CAS號:120068-37-3)、氟硅唑(Fflusilazole, CAS號:85509-19-9)、丙環唑(propiconazole, CAS號:60207-90-1)、炔螨特(propargite, CAS號:2312-35-8)、甲氰菊酯(fenpropathrin, CAS號:39515-41-8)、雙甲脒(amitraz, CAS號:33089-61-1)、氯氟氰菊酯(cyhalothrin, CAS號:68085-85-8)、噠螨靈(pyridaben, CAS號:96489-71-3)、氰戊菊酯(fenvalerate, CAS號:51630-58-1)、順式氰戊菊酯(es-fenvalerate, CAS號:66230-04-4)、苯醚甲環唑(difenoconazole, CAS號:119446-68-3)、氯氰菊酯(cypermethrin, CAS號:52315-07-8)20種標準品,質量濃度均為100 mg/L標準溶液(農業部環境保護科研監測所);乙腈、丙酮、正己烷均為色譜純(賽默飛世爾科技(中國)有限公司);吸附劑C18、GCB、PSA(上海安譜實驗科技股份有限公司)。
1.2 色譜條件與質譜條件
色譜柱為DB-5MS UI石英毛細管色譜柱,30 m×0.25 mm(內徑)×0.25 μm(膜厚)。色譜柱升溫程序:40 ℃下保持1 min,以30 ℃/min的速率升至130 ℃,以5 ℃/min的速率升至250 ℃,以10 ℃/min的速率升至300 ℃,保持15 min。進樣體積為1 μL;進樣方式為不分流進樣。
離子源為電子轟擊(EI)源;數據采集模式為MRM模式。進樣口溫度為290 ℃,離子源溫度為230 ℃,四極桿(Q1和Q3)溫度為150 ℃,輔助溫度為280 ℃。載氣為純度≥99.999%的氦氣,流速為1.0 mL/min;碰撞氣為氮氣,電離能量為70 eV。定量、定性離子對和碰撞能量參數具體見表1。
1.3 樣品的前處理
稱取3.00 g已粉碎試樣于50 mL的聚四氟乙烯離心管中,加10 mL乙腈,均質提取1 min,加4 g無水硫酸鎂和1 g氯化鈉,漩渦振蕩1 min,以4 500 r/min的速度離心5 min,取上清液待用。另取一支15 mL聚四氟乙烯離心管,加入1 200 mg無水硫酸鎂、45 mg GCB、400 mg C18和400 mg PSA粉吸附劑,加入6 mL上述上清液,渦旋混合1 min,以4 500 r/min的速度離心5 min,移取2 mL于15 mL試管中,氮吹濃縮近干,用丙酮-正己烷(3∶7, v/v)混合溶液定容至2 mL,待上機測試。
2 結果與討論
2.1 質譜條件優化
參考GB 23200.8-2016中檢測條件,優化質譜條件。首先將20種農藥分別配制成1 mg/L的單標準溶液,按照全掃描方式采集某種農藥單標準溶液,選擇豐度高、質荷比大且選擇對稱性好的離子作為母離子,然后在3~45 eV的范圍內,每3 eV為間隔,按照產物離子掃描方式優化碰撞電壓,選擇強度和信噪比兼顧的離子分別作為定量和輔助定性子離子,最終得到優化后的質譜參數:母離子、子離子、碰撞電壓(見表1)。
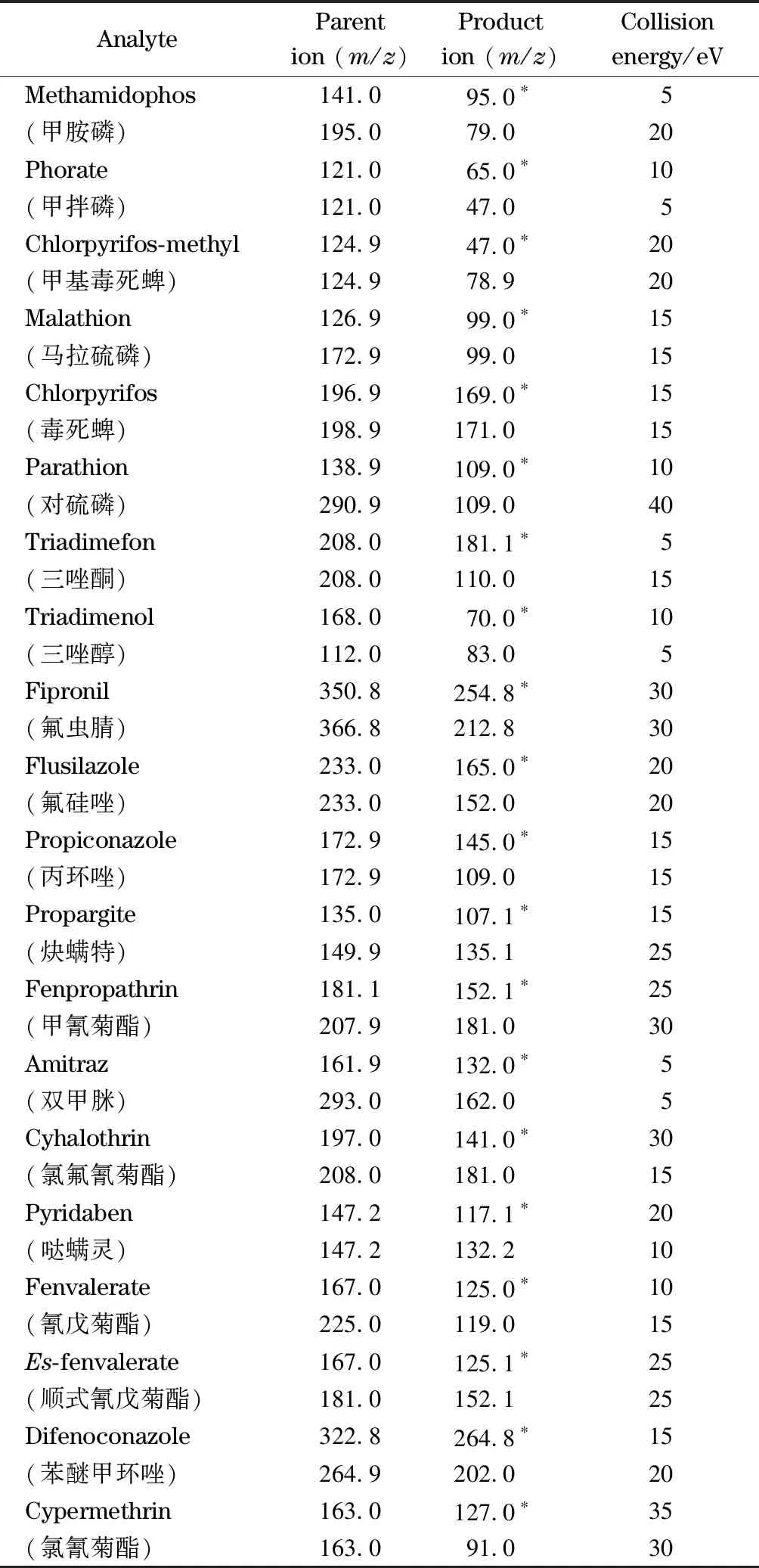
表 1 20種農藥的定量、定性離子對和碰撞能量
* Quantitative ion.
按表1中條件對20種農藥的基質匹配混合標準溶液進行MRM采集,得到其總離子流圖,見圖1。
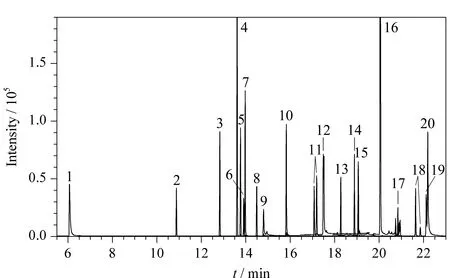
圖 1 質量濃度均為100 μg/L的20種農藥的基質匹配混合標準溶液在MRM模式下的總離子流圖Fig. 1 Total ion chromatogram for a mixed matrix- matched standard solution of the 20 pesticides (100 μg/L for each) in MRM mode Peak Nos.: 1. methamidophos; 2. phorate; 3. chlorpyrifos-methyl; 4. malathion; 5. chlorpyrifos; 6. parathion; 7. triadimefon; 8. fipronil; 9. triadimenol; 10. flusilazole; 11. propiconazole; 12. propargite; 13. fenpropathrin; 14. cyhalothrin; 15. amitraz; 16. pyridaben; 17. cypermethrin; 18. fenvalerate; 19. es-fenvalerate; 20. difenoconazole.
2.2 提取溶劑的選擇
在農藥殘留分析中,一般用甲醇、丙酮、乙腈、乙酸乙酯、正己烷、二氯甲烷等作為提取溶劑,而二氯甲烷或二氯甲烷-石油醚毒性很大,對實驗人員構成健康威脅。本試驗考察了丙酮、正己烷、乙腈3種溶劑對樣品的提取效果。設定添加水平為2倍定量限(LOQ),其余步驟按照1.3節進行試驗,平行檢測3次,結果表明,丙酮提取出的雜質和色素較多;正己烷很難將極性大的農藥提取出來,而極性小的油脂易被共萃出來;而乙腈提取效果最好,對農藥的溶解性較強,對油脂和色素等雜質溶解度較小,提取效率最佳。
2.3 提取溶劑使用量及提取時間優化
提取溶劑使用量及提取時間的選擇直接關系到測定結果的回收率。本文以乙腈作為提取溶劑,考察了二者對回收率的影響(見圖2)。當提取時間超過2 min時,回收率基本變化不大;提取溶劑用量為14 mL時,回收率最高,但用量為10 mL時,提取效率也可以滿足試驗要求。綜合考慮了提取效率、基質效應、經濟效益等方面,最終確定乙腈的用量為10 mL,提取時間為2 min。
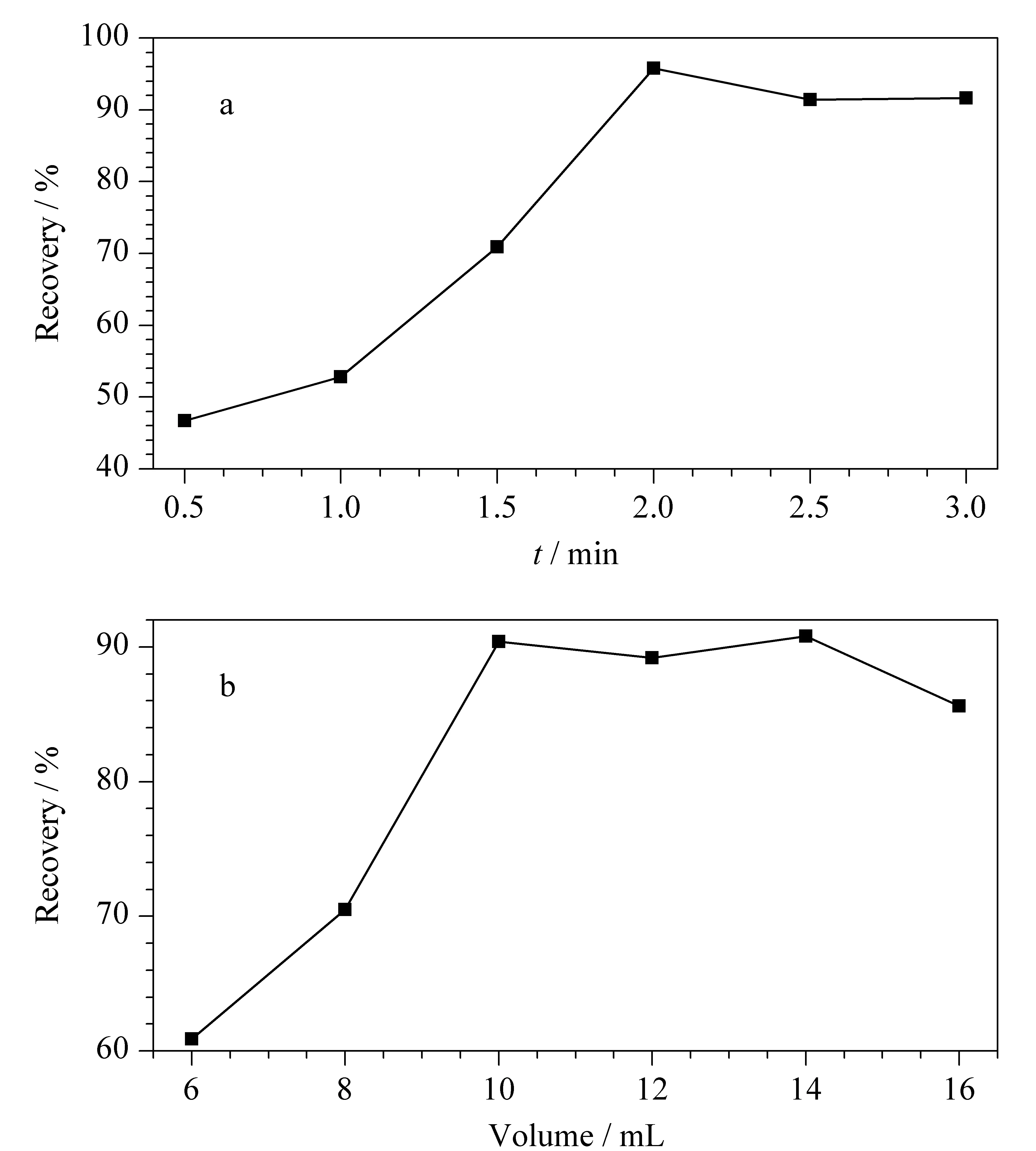
圖 2 (a)提取時間和(b)乙腈用量對20種農藥回收率的影響Fig. 2 Effects of (a) extraction time and (b) addition amount of acetonitrile on the recoveries of the 20 pesticides
2.4 吸附劑種類的選擇
在進一步凈化的過程中,選擇C18、GCB和PSA作為分散固相萃取吸附劑,比較了不同吸附劑對目標物凈化回收效果的影響。C18是一種非極性的廣譜性凈化吸附劑,以C18為分散固相萃取吸附劑時,雖然樣品提取溶液顏色較深,但其能夠有效去除提取物中微量的非極性雜質。GCB吸附劑表面具有正六元環結構,使其對平面分子有極強的親和力,能夠分離或去除枸杞中的色素以及固醇類雜質,有效去除樣品提取液的顏色。PSA吸附劑結構中含有兩個氨基,通過離子交換作用或者氫鍵的形成,能有效去除樣品中的有機脂肪酸和糖等干擾物[19],這種混合型分散固相萃取吸附劑可以明顯改善凈化效果,因此,本試驗選擇PSA、GCB和C18混合物作為分散固相萃取吸附劑進行考察。當PSA使用量為400 mg時,凈化效果良好,定性定量更加準確;低于400 mg時,凈化不完全,干擾較為嚴重;大于400 mg時,其吸附農藥的量會隨之增加,影響農藥的測定。當C18加入量為400 mg時,20種農藥的回收率較高;當加入量低于或高于400 mg時,回收率較低。加入GCB的量在45 mg為宜,脫色效果最佳。綜合考慮,本試驗采用45 mg GCB, 400 mg C18和400 mg PSA粉作為吸附劑。
2.5 靈敏度考察
由于枸杞樣品中含有較多的糖類、油脂、色素等復雜雜質,可能對檢測結果造成影響,鑒于此,分別采用SIM和MRM模式進行考察。以馬拉硫磷、毒死蜱、對硫磷作為目標物,制備枸杞基質加標樣品,分別進行SIM和MRM兩種模式掃描,結果見圖3。
SIM模式針對一級質譜而言,即只掃描一個特征離子,目標離子響應高,但基質噪音也大。而MRM模式針對二級質譜而言,Q1和Q3分別采集一個特征離子,進一步降低噪音和基質干擾,信噪比會更高,定性更準確,尤其對于復雜基質、背景高的樣品。通過圖3的比較可知,SIM模式下的色譜圖背景噪音高,信號干擾大;MRM模式采集譜圖的背景噪音明顯降低,S/N升高。因此MRM模式較SIM模式靈敏度更高,定性更準確。
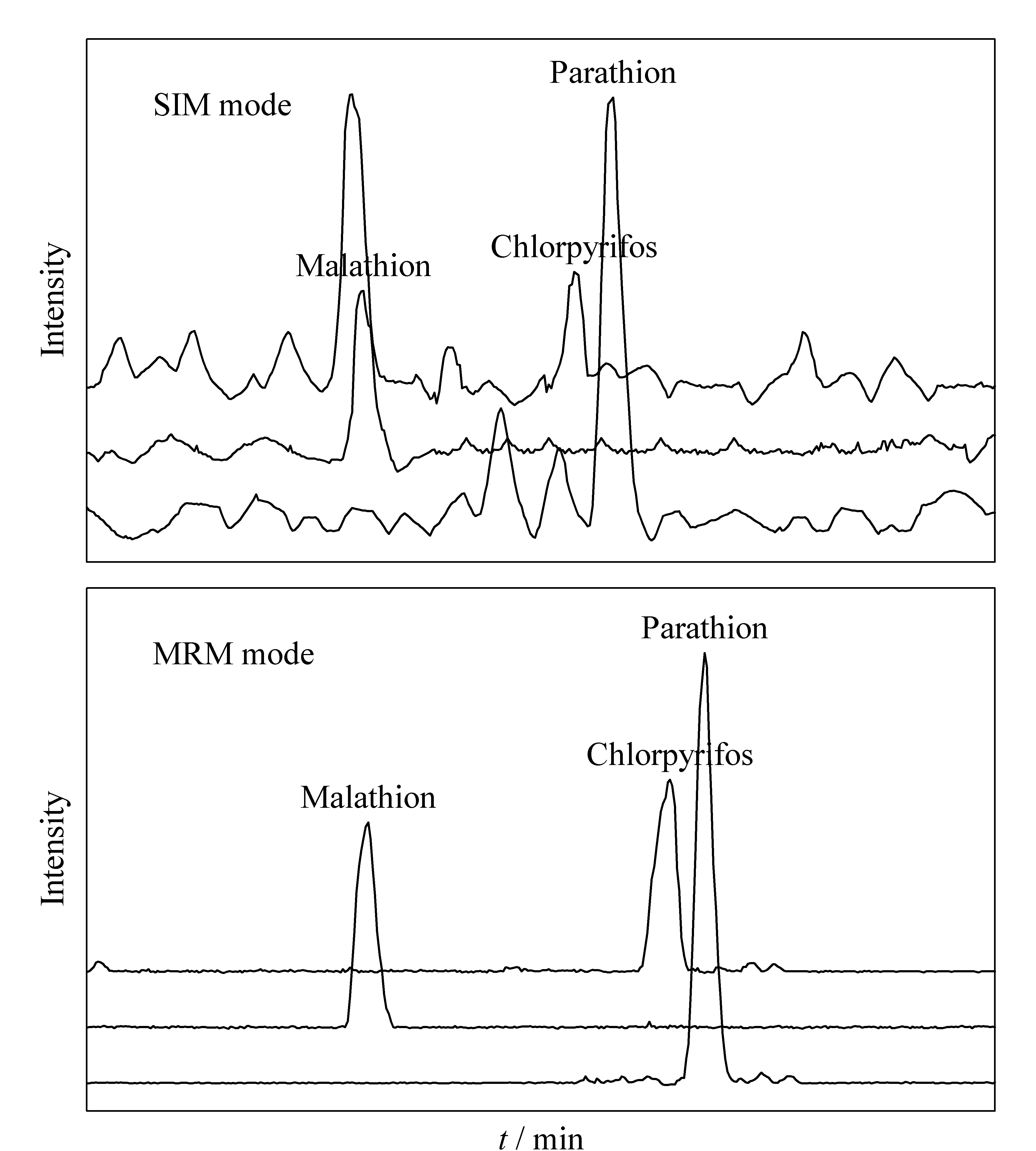
圖 3 枸杞基質加標溶液(馬拉硫磷、毒死蜱和對硫磷)在SIM和MRM模式下的色譜圖Fig. 3 Chromatograms of wolfberry matrix spiked with standard solutions of malathion, chlorpyrifos, and parathion in SIM mode and MRM mode
2.6 基質效應考察
基質效應是指色譜分離時共洗脫的物質改變了待測成分的離子化效應,所引起的信號的抑制或提高。基質效應影響大時會降低方法的靈敏度和準確性,給測定帶來誤差,本文通過配制標準工作曲線和基質匹配校正曲線,以峰面積為縱坐標,質量濃度為橫坐標作圖,以基質匹配校正曲線和標準曲線斜率的比值來考察化合物的基質效應。一般認為,基質匹配校正曲線斜率與標準曲線斜率的比值在85%~115%之間不存在基質效應[20-23]。圖4表明,對于甲胺磷、甲拌磷、甲基毒死蜱、毒死蜱、三唑酮、三唑醇峰2、氟硅唑、丙環唑、炔螨特、甲氰菊酯、雙甲脒、噠螨靈、氯氰菊酯,其基質匹配校正曲線斜率為標準曲線斜率的74.1%~111.8%,不存在基質效應;而對于馬拉硫磷、對硫磷、三唑醇峰1、氟蟲腈、氯氟氰菊酯峰、氰戊菊酯和苯醚甲環唑峰,基質匹配校正曲線斜率分別為標準曲線斜率的118.6%~135.2%,存在明顯的基質增強作用。綜合考慮,本文采用基質匹配標準溶液校準方法進行準確定量分析。
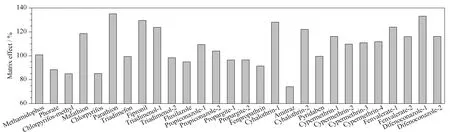
圖 4 20種農藥在枸杞樣品中的基質效應Fig. 4 Matrix effects for the 20 pesticides in wolfberry
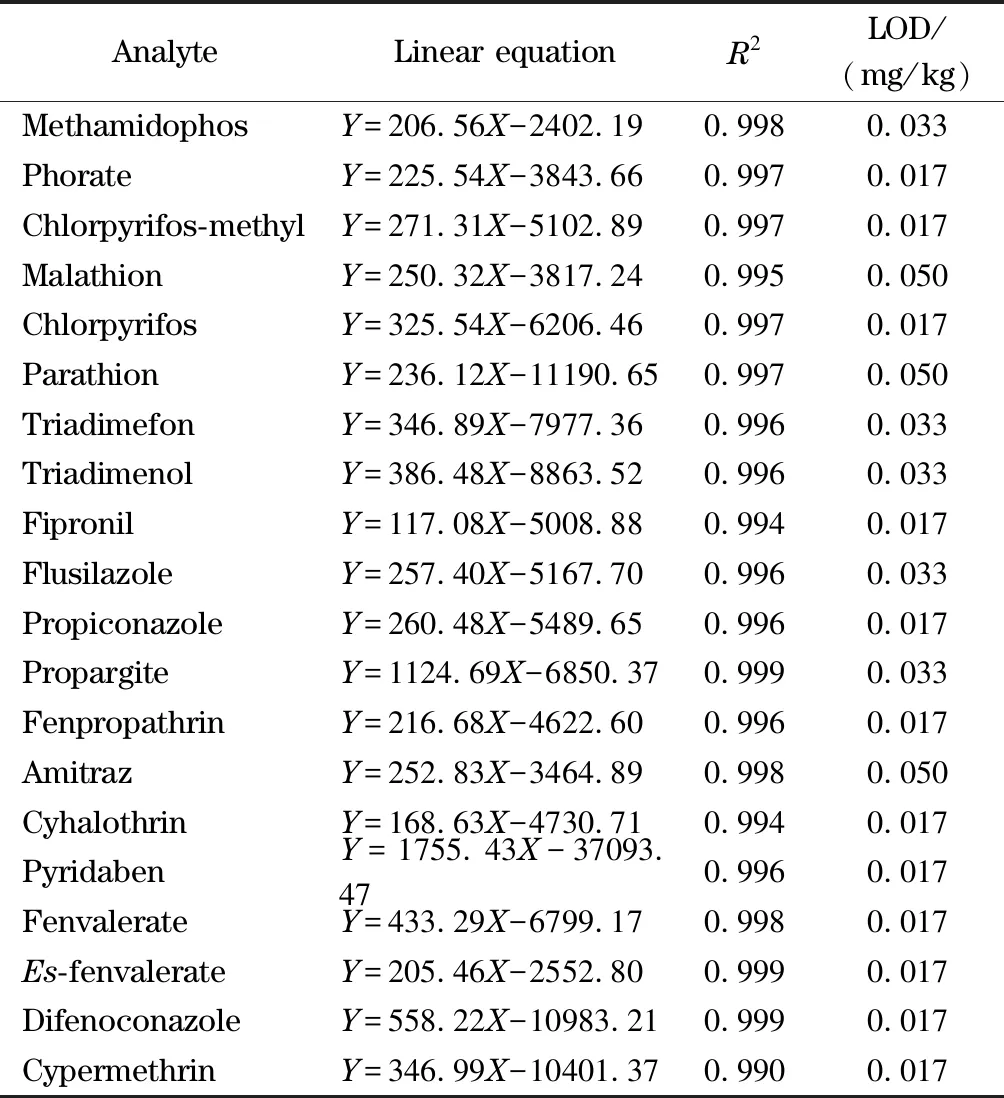
表 2 20種農藥的線性方程、相關系數及檢出限
Y: peak area;X: mass concentration, μg/L.
2.7 標準曲線與檢出限
由于基質效應的存在,使用基質匹配標準樣品進行定量分析。配制質量濃度水平分別為5、10、25、50、100和500 μg/L的20種農藥溶液,建立20種農藥在枸杞基質中的標準曲線。所有農藥的線性良好,線性相關系數(R2)均不小于0.99,各農藥的線性相關系數在0.990~0.999之間。
稱取20份枸杞,分別在其中添加某一預期濃度點的標準品,按照1.3節步驟進行前處理,然后上機測試,保證每次均有響應信號,且S/N在3附近,那么該濃度即為此種農藥的方法檢出限。
線性方程、相關系數及檢出限見表2。檢出限為0.017~0.050 mg/kg,以檢出限的3倍確定定量限。方法線性范圍可以滿足枸杞樣品檢測的需要。
2.8 方法精密度和回收率
設定添加水平為1、2、10倍LOQ,分別稱取6份枸杞試樣,按照1.3節步驟進行試驗,其回收率在71.5%~109.7%之間,測定結果的相對標準偏差在1.94%~9.87%之間,詳見表3。
2.9 方法應用
本方法具有快速、簡便、可操作性強、準確度高和精密度好的優點,可應用于枸杞樣品中多種農藥殘留的篩查。選取20個枸杞樣本,應用本方法進行了分析測試,檢測結果顯示本地區枸杞中氯氟氰菊酯、噠螨靈、苯醚甲環唑和氯氰菊酯檢出率分別為30%、25%、35%、30%,其中氯氟氰菊酯檢出值達1.759 mg/kg,以上4種農藥風險系數較高。在今后枸杞種植過程中應嚴格控制農藥種類及劑量使用,加強枸杞產品質量安全建設,以滿足出口國家或地區殘留限量的要求,從而保護消費者的身體健康。
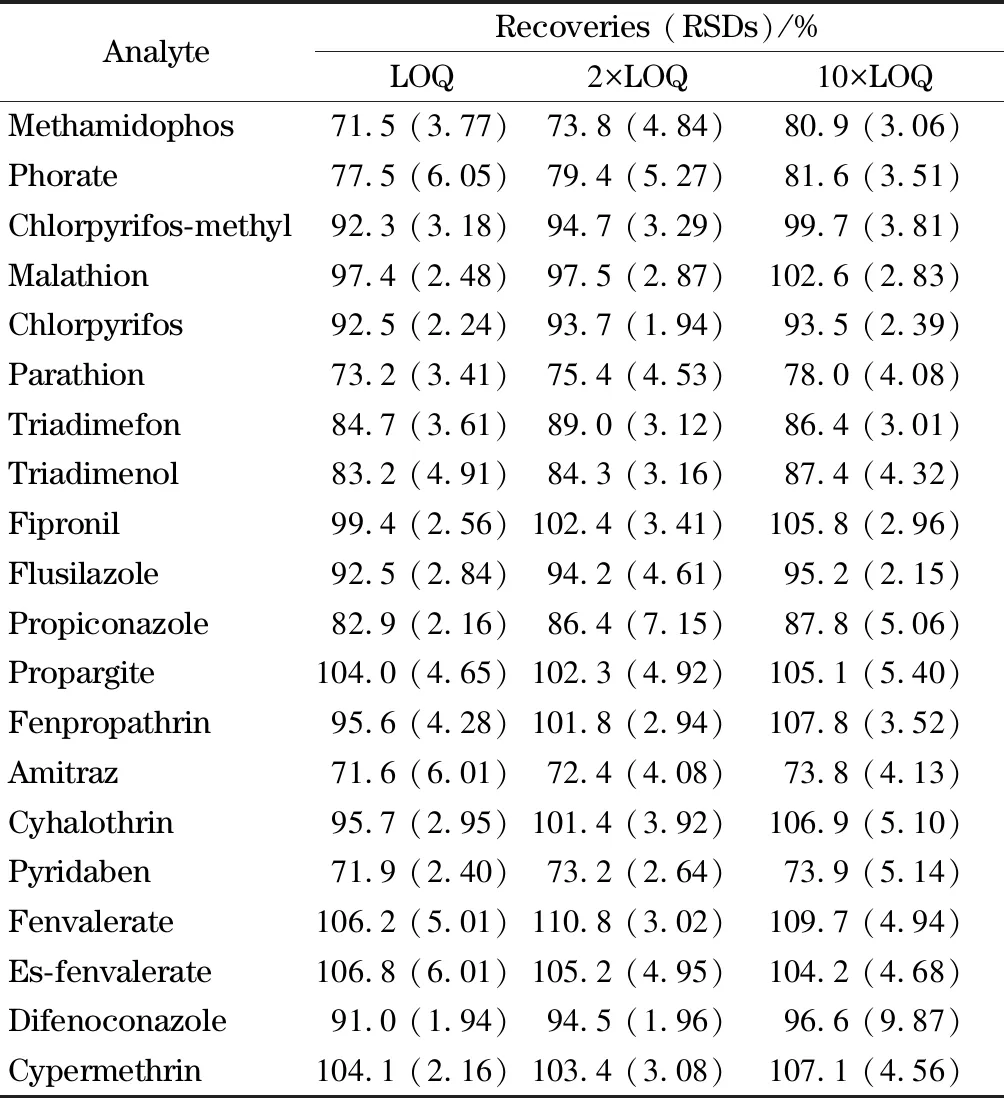
表 3 20種農藥在枸杞中3個添加水平下的加標回收率
3 結論
本文建立了QuEChERS-氣相色譜-串聯質譜法測定枸杞中多種農藥殘留,前處理方法省時省力,操作簡便,不易交叉污染,MRM模式減少了基質對農藥的定量、定性離子干擾,同時也提高了方法的靈敏度。該方法分離效果好、準確度高、重復性好,在實際工作中取得了令人滿意的結果。
