分散固相萃取法結(jié)合UPLC-MS/MS同時(shí)分析茶葉中13種殺菌劑
2019-09-12 07:39:54劉文靜吳建鴻
福建茶葉 2019年5期
關(guān)鍵詞:檢測
黃 彪,劉文靜,吳建鴻
(福建省農(nóng)業(yè)科學(xué)院農(nóng)業(yè)質(zhì)量標(biāo)準(zhǔn)與檢測技術(shù)研究所/福建省農(nóng)產(chǎn)品質(zhì)量安全重點(diǎn)實(shí)驗(yàn)室,福建福州 350003)
1 前言
在農(nóng)業(yè)領(lǐng)域,殺菌劑主要是指用于防治由各種病原微生物引起的植物病害的一類農(nóng)藥。殺菌劑能很好的防治植物病害,起到保護(hù)植物、促進(jìn)生產(chǎn)的作用,被廣泛應(yīng)用于農(nóng)作物的種植過程。由于農(nóng)藥殘留的潛在危害,許多國家及國際組織都對(duì)農(nóng)藥殘留嚴(yán)格規(guī)定了殘留限量[1-2]。我國茶葉品種資源多,種植區(qū)域廣,茶樹在種植過程中從葉、莖、根到花等均可能有病蟲危害,因此殺菌劑在茶葉的種植過程中應(yīng)用較多,國際貿(mào)易中對(duì)農(nóng)產(chǎn)品中農(nóng)藥殘留最高限量的規(guī)定越來越嚴(yán)格,有關(guān)茶葉的農(nóng)藥殘留限量標(biāo)準(zhǔn)也更為嚴(yán)苛,而我國一直以來是重要的茶葉出口大國,因此研究茶葉中殺菌劑殘留的檢測方法具有非常重要的意義。
快速、高效的農(nóng)藥殘留分析方法能否建立在很大程度上取決于樣品前處理過程,高效的前處理技術(shù)往往能大大減少分析的成本和時(shí)間。目前,農(nóng)產(chǎn)品中殺菌劑的檢測常見前處理方法有液液萃取、固相柱萃取、分子印跡固相萃取及分散固相萃取等[3-7]。分散固相萃取技術(shù)相比于其他前處理方法具有溶劑用量少,提取速度快,重現(xiàn)性好等優(yōu)點(diǎn),在樣品前處理過程中得到廣泛應(yīng)用。殺菌劑的檢測方法主要有毛細(xì)管電泳法[8]、氣相色譜法[9-10]、液相色譜法[11-12]、氣相色譜-串聯(lián)質(zhì)譜法[13-14]、液相色譜-串聯(lián)質(zhì)譜法[15-17]等。
質(zhì)譜檢測技術(shù)因其抗干擾強(qiáng),檢測快速,靈敏度高,定性定量準(zhǔn)確等優(yōu)點(diǎn),已逐漸發(fā)展成為分析檢測中的主導(dǎo)技術(shù),而尤其是超高效液相色譜-串聯(lián)質(zhì)譜分析技術(shù),將超高效液相色譜儀對(duì)復(fù)雜樣品的快速分離能力與質(zhì)譜儀對(duì)目標(biāo)離子的高選擇性、高靈敏度相結(jié)合,在食品安全、藥物分析、環(huán)境監(jiān)測等領(lǐng)域中越來越發(fā)揮出重要作用。在已有報(bào)道中,利用分散固相萃取結(jié)合超高效液相色譜-串聯(lián)質(zhì)譜法檢測茶葉中殺菌劑殘留的報(bào)道尚不多見。本研究利用分散固相萃取法前處理凈化,建立了超液相色譜-串聯(lián)質(zhì)譜法分析茶葉中殺菌劑殘留。該方法樣品前處理時(shí)間短,分析快速,準(zhǔn)確性好,可為茶葉殺菌劑殘留定性定量分析提供方法參考依據(jù)。
2 材料與方法
2.1 儀器與試劑
ACQUITY UPLC H-Class超高效液相色譜儀(美國Waters公司);Xevo TQ-S三重四極桿質(zhì)譜儀(美國Waters公司);樣品均質(zhì)器(德國IKA公司);SYG-2水浴恒溫振蕩器(常州朗越儀器有限公司);XW-80A旋渦混合器(上海醫(yī)科大學(xué)儀器廠);Millipore Qirect-Q5超純水設(shè)備(美國Millipore公司)。
乙腈(色譜純,美國Waters公司)、甲醇(色譜純,美國Waters公司)、乙酸銨(LC-MS級(jí),美國 Waters公司)、甲酸(LC-MS級(jí),美國Waters公司)、N-丙基乙二胺(PSA,40 μm,天津博納艾杰爾科技有限公司)、C18吸附劑(40-60 μm,天津博納艾杰爾有限公司)、超純水(Millipore超純水機(jī)制備),其他試劑為分析純。
2.2 標(biāo)準(zhǔn)溶液的配制
氟硅唑、丙環(huán)唑標(biāo)準(zhǔn)品(純度97%,德國DR公司),用乙腈溶解配制成濃度為1 000 mg/L的標(biāo)準(zhǔn)儲(chǔ)備液;三唑酮、苯醚甲環(huán)唑、多效唑等農(nóng)藥標(biāo)準(zhǔn)液購于國家標(biāo)準(zhǔn)物質(zhì)中心,濃度規(guī)格為1 000 mg/L。標(biāo)準(zhǔn)品儲(chǔ)備液置于-20℃冰箱中保存,實(shí)驗(yàn)時(shí)取儲(chǔ)備液配制成10 mg/L的單標(biāo)中間液,用空白基質(zhì)溶液配制的用于制作標(biāo)準(zhǔn)曲線的不同濃度的混標(biāo)工作液,現(xiàn)配現(xiàn)用。
2.3 樣品前處理方法
準(zhǔn)確稱取5.00 g(精確至0.01 g)磨碎的茶葉樣品于50 mL離心管中,加入10 mL乙腈,用均質(zhì)器以15000 r/min均質(zhì)提取1 min后,蓋好蓋子振蕩提取15min,以 8 000 r/min離心5 min;轉(zhuǎn)移上清液2.0 mL到預(yù)先加有50 mg PSA,50 mg C18,10 mg石墨化炭黑GCB的10 mL離心管中,渦旋30 s后,8 000 r/min離心1min;取上清液過0.22 μm 濾膜,濾液供UPLC-MS/MS分析。
2.4 色譜-質(zhì)譜條件
超高效液相色譜條件:Waters T3 C18色譜柱(2.1 mm×50 mm,1.8 μm,);柱溫:30.0℃;梯度洗脫:流動(dòng)相A為0.1%甲酸-5 mmol/L乙酸銨水溶液,流動(dòng)相 B 為乙腈;0~3 min,90%~70%A ;3~5 min,維持 70%A;5~8 min,70%~20%A;8~10 min,20%~90%A;流動(dòng)相流速:0.2 mL/min;進(jìn)樣量:4 μL。
質(zhì)譜條件:電噴霧ESI+掃描方式;檢測方式:多反應(yīng)監(jiān)測(MRM)模式;毛細(xì)管電壓:2.5 kV;錐孔氣流:氮?dú)猓魉?0 L/h;脫溶劑溫度:150℃,流速 800 L/h;離子源溫度:150 ℃;霧化氣壓力:0.28 MPa;碰撞氣:高純氬氣。相關(guān)質(zhì)譜檢測參數(shù)見表1。
3 結(jié)果與討論
3.1 色譜-質(zhì)譜條件的優(yōu)化
在實(shí)驗(yàn)過程中,比較了以甲醇-水和乙腈-水作為流動(dòng)相以及在流動(dòng)相中添加甲酸、乙酸銨對(duì)目標(biāo)化合物的分離效果及目標(biāo)離子色譜峰峰型的影響。實(shí)驗(yàn)結(jié)果表明:采用乙腈-5 mmol/L乙酸銨水溶液(含0.1%甲酸)作為流動(dòng)相時(shí),在正離子掃描模式下,殺菌劑有良好的峰形且相對(duì)于不加甲酸的流動(dòng)相有更好的響應(yīng)強(qiáng)度,甲酸的加入有利于目標(biāo)分子更好的形成正離子,5 mmol/L乙酸銨-0.1%甲酸水溶液緩沖體系在一定程度上可以調(diào)節(jié)體系的pH值,使得在一個(gè)合適的酸堿性范圍內(nèi)目標(biāo)離子有更好的峰型。
3.2 提取溶劑的選擇
在農(nóng)藥目標(biāo)分子的前處理過程中,常用的萃取溶劑有甲醇、乙腈、乙酸乙酯等。甲醇雖對(duì)農(nóng)藥分子有很好的溶解性,但甲醇是一種質(zhì)子性溶劑在提取農(nóng)藥分子的同時(shí)也很容易將樣品中的醇類、多酚、有機(jī)酸等物質(zhì)提取出來。茶葉樣品中既有較多的茶多酚等物質(zhì),同時(shí)也含有沒食子酸等一些有機(jī)酸物質(zhì),采用甲醇作為提取茶葉樣品中的農(nóng)藥分子,會(huì)對(duì)后續(xù)的凈化過程造成一定的影響。乙睛作為一種極性較大的溶劑、穿透力強(qiáng),對(duì)農(nóng)藥分子的提取效率高,并且用乙腈作為提取溶劑可以減少維生素、色素等中等極性雜質(zhì)的溶出,因此被廣泛用于殘留分析中目標(biāo)分子的提取。如用乙酸乙酯作為提取溶劑,最后需氮吹轉(zhuǎn)溶劑為乙腈或者甲醇進(jìn)行液相色譜-質(zhì)譜分析,會(huì)使得前處理時(shí)間變長。因此綜合考慮,采取乙腈作為提取溶劑。

表1 13種殺菌劑檢測的質(zhì)譜參數(shù)
3.3 分散固相萃取劑的選擇
在樣品前處理過程中常用的固相萃取劑有PSA、C18及GCB。PSA是含有氨基基團(tuán)的強(qiáng)極性分子,能夠通過強(qiáng)的吸附及離子交換過程將與基質(zhì)中的多酚、有機(jī)酸、糖類等含有羥基或者羧基的極性分子除去。C18由于分子中鍵合十八烷基,對(duì)極性較小的分子具有良好的吸附作用,能夠去除基質(zhì)中的色素、維生素等物質(zhì)。GCB則對(duì)色素有良好的去除能力。考慮到茶葉基質(zhì)中有茶多酚、有機(jī)酸、維生素及色素等物質(zhì),因此在實(shí)驗(yàn)中考慮將上述吸附劑進(jìn)行組合對(duì)樣品前處理凈化。在實(shí)驗(yàn)中以茶葉樣品為基質(zhì)加入50 μg/kg混標(biāo)溶液,考察了不同用量固相萃取劑的凈化效果及回收率情況:(1)25 mg PSA+25 mg C18+10 mg GCB(2)50 mg PSA+50 mg C18+10 mg GCB(3)75 mg PSA+75 mg C18+10 mg GCB。實(shí)驗(yàn)結(jié)果表明:以乙睛為提取溶劑的情況下,第(2)、(3)兩種吸附劑綜合使用效果都較好,能夠較好的對(duì)樣品前處理去除雜質(zhì),并且目標(biāo)分子的回收率均在75%以上。另外也考察了PSA和C18用量不變時(shí),改變GCB用量凈化效果的不同:(4)50 mg PSA+50 mg C18+5 mg GCB (5)50 mg PSA+50 mg C18+10 mg GCB(6)50 mg PSA+50 mg C18+15 mg GCB。實(shí)驗(yàn)中發(fā)現(xiàn),加入 10 mg GCB即可很好的去除色素等雜質(zhì)。因此,綜合考慮實(shí)驗(yàn)成本、凈化效以及對(duì)回收率的影響,實(shí)驗(yàn)中采用50 mg PSA+50 mg C18+10 mg GCB吸附劑組合進(jìn)行樣品的凈化處理。
3.4 方法評(píng)價(jià)
將空白樣品經(jīng)固相萃取劑分散提取得到基質(zhì)凈化液,配制13種殺菌劑的基質(zhì)匹配混標(biāo)溶液,注入超高效液相色譜-串聯(lián)質(zhì)譜儀進(jìn)行分析。以目標(biāo)物特征離子色譜峰響應(yīng)信號(hào)峰面積(y)為縱坐標(biāo),目標(biāo)化合物的基質(zhì)匹配溶液濃度(μg/L)x為橫坐標(biāo),制作標(biāo)準(zhǔn)曲線。依據(jù)特征離子色譜峰信號(hào)的3倍信噪比(S/N≥3)確定方法的檢出限,根據(jù)各目標(biāo)化合物的響應(yīng)情況在茶葉基質(zhì)中進(jìn)行低、中、高3個(gè)濃度水平的加標(biāo)。結(jié)果表明:13殺菌在0.5 μg/L~100 μg/L濃度范圍內(nèi)基質(zhì)匹配曲線線性關(guān)系良好(r≥0.995),方法靈敏度高,目標(biāo)化合物的檢出下限在 0.08~0.7 μg/kg之間。不同濃度水平加標(biāo)相應(yīng)平均回收率為70.1%~106.7%,相對(duì)標(biāo)準(zhǔn)偏差在 1.2%~7.6%之間(表 2)。
4 實(shí)際樣品的檢測
采集茶葉樣品40余份進(jìn)行了殺菌劑殘留的分析,發(fā)現(xiàn)1份茶葉樣品中有苯醚甲環(huán)唑殘留,含量為0.056 mg/kg,陽性樣品的MRM離子色譜圖如圖1所示。雖有檢出,但未超出食品安全國家標(biāo)準(zhǔn)限量規(guī)定(標(biāo)準(zhǔn)規(guī)定茶葉中苯醚甲環(huán)唑最大殘留限量為10 mg/kg)[2]。
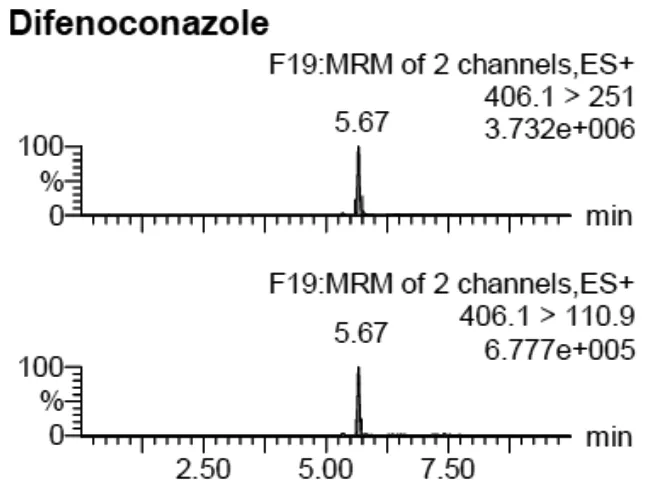
圖1 陽性樣品的苯醚甲環(huán)唑MRM離子色譜圖

表2 13種殺菌劑的平均加標(biāo)回收率和檢出限(n=6)
5 結(jié)論
本研究運(yùn)用分散固相萃取前處理法結(jié)合超高效液相色譜-串聯(lián)質(zhì)譜法,建立了茶葉中多菌靈、三唑酮、腈菌唑、苯醚甲環(huán)唑等13種殺菌劑殘留的檢測方法。該方法前處理簡單,方便快速,具有良好的靈敏度、準(zhǔn)確度和精密度,可用于茶葉中殺菌劑殘留的同時(shí)分析,有利于農(nóng)產(chǎn)品質(zhì)量安全的保障。
猜你喜歡
中國設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
