新型三維磁性豆甾醇印跡聚合物制備及其性能研究
2019-09-10 09:36:44閆亮丁旺莎楊仕锃王兆玉李嘉豪張月
現(xiàn)代鹽化工 2019年6期
閆亮 丁旺莎 楊仕锃 王兆玉 李嘉豪 張月



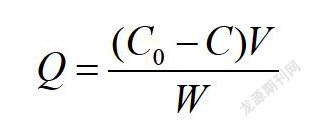
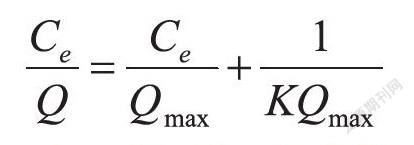
摘? ?要:以多壁碳納米管和氧化石墨烯三維磁性復(fù)合材料為載體,豆甾醇為模板分子,多巴胺為功能單體,制備新型三維磁性豆甾醇印跡復(fù)合萃取材料。采用掃描電鏡、紅外光譜和X射線衍射對該復(fù)合材料進(jìn)行表征和分析,結(jié)果表明:在三維磁性材料表面成功接枝印跡層。采用紫外可見光譜儀對該印跡復(fù)合材料的吸附性能進(jìn)行探討,結(jié)果表明:三維磁性印跡復(fù)合材料對豆甾醇表現(xiàn)出特異性吸附性能,最大吸附量為11.24? mg/g。
關(guān)鍵詞:石墨烯;碳納米管;豆甾醇;三維磁性材料;分子印跡
分子印跡是指制備對某一特定的目標(biāo)分子具有特異選擇性的分子印跡聚合物(Molecular Imprinting Polymer,MIPs)的技術(shù)[1]。因其對目標(biāo)分子具有特異選擇性識別能力,MIPs已廣泛用于萃取分離、化學(xué)/生物傳感和人工抗體等領(lǐng)域[2]。最近,有科研工作者以多壁碳納米管(Multi-Walled Carbon Nanotube,MWNTs)為基材,結(jié)合印跡技術(shù),制備具有高選擇性和高吸附容量的碳納米管-分子印跡聚合物(MWNTs-MIPs)。
新型磁性三維印跡復(fù)合材料借助碳納米管和石墨烯的比表面積優(yōu)勢,制備出具有更高的吸附容量的三維印跡萃取材料[3]。但是目前的磁性碳基三維基質(zhì)材料在制備過程中還有結(jié)構(gòu)不穩(wěn)定和制備能耗較大的問題,限制了分離領(lǐng)域的使用[4]。最近有報道采用水熱法制備磁性碳基三維印跡納米復(fù)合材料,這種水熱法制備的復(fù)合材料具有較強(qiáng)的穩(wěn)定性,為新型磁性碳基表面三維印跡納米復(fù)合材料的廣泛應(yīng)用提供了可能性。
植物甾醇具有降低血清膽固醇、消炎、退熱和抗癌等藥理功效,也是活性成分研究中的重要對象。本研究采用石墨烯/碳納米管三維復(fù)合材料為支撐基質(zhì),結(jié)合納米表面印跡技術(shù),在溫和條件下研制出高吸附性能和快速吸附的石墨烯/碳納米管三維質(zhì)表面豆甾醇印跡復(fù)合萃取材料,并對其吸附性能進(jìn)行研究。
1? ? 實(shí)驗(yàn)部分
1.1? 試劑與儀器
多壁碳納米管(直徑為20~50 nm,純度大于95%)購自深圳比爾科技公司;石墨烯(GO)、豆甾醇(Stig)、β-谷甾醇(β-Sito)、膽固醇(Chol)、三羥甲基氨基甲烷(Tris)、多巴胺購自阿拉丁公司;硫酸、硝酸、乙酸、FeCl3?6H2O、高錳酸鉀、5%過氧化氫溶液、檸檬酸鈉、肼購自長沙化學(xué)試劑公司。除特殊說明外,所用試劑均為分析純;實(shí)驗(yàn)用水為二次蒸餾水。
Zeiss Sigma HD掃描電鏡(德國蔡司公司),F(xiàn)T-IR傅里葉紅外光譜儀(日本島津公司),高效液相色譜儀(日本島津公司),DZF-6020型真空干燥箱(上海精宏實(shí)驗(yàn)設(shè)備有限公司)。
1.2? 實(shí)驗(yàn)過程
1.2.1? 三維磁性復(fù)合材料制備
將1.0 g多壁碳納米管加入20.0 mL 65%硝酸和60.0 mL 98%硫酸混合溶液中,超聲1 h,升溫至80 ℃反應(yīng)2 h,過濾得到活化的碳納米管。再將制得的200.0 mg GO和200.0 mg活化碳納米管分散在160.0 mL去離子水中超聲3 h。將3.2 g FeCl3·6H2O和5.9 g檸檬酸鈉分別加入到40.0 mL和80.0 mL去離子水中,在磁力攪拌下逐滴加入上述混合液,再加入12.0 mL肼繼續(xù)攪拌30 min。將混合物轉(zhuǎn)移至100.0 mL反應(yīng)釜中,180 ℃下反應(yīng)12 h。采用0.45 μm薄膜過濾,用去離子水和乙醇各洗滌3次,80 ℃真空干燥24 h,得最終產(chǎn)物。
1.2.2? 三維磁性印跡復(fù)合材料制備
取400.0 mg三維復(fù)合材料和400.0 mL Tris緩沖液于圓底燒瓶中,超聲5 min,加入100.0 mg豆甾醇和360.0 mg多巴胺。機(jī)械攪拌8 h,將產(chǎn)物過濾,先用水洗滌3次,然后用乙酸/甲醇(體積比為2∶8)混合液反復(fù)洗滌,接著用乙醇反復(fù)沖洗至收集的洗脫液在豆甾醇溶液標(biāo)準(zhǔn)紫外峰處沒有明顯的吸收。真空干燥12 h,制備出三維磁性印跡復(fù)合材料(3D-MMIPs)。
在不加入豆甾醇的前提下,采用同樣的方法,制備出三維磁性非印跡復(fù)合材料(3D-MNIPs)。
1.3? 吸附性能研究
1.3.1? 動態(tài)吸附過程
取20 mg 3D-MMIPs 或3D-MNIPs分別置于吸附管中,加入10.0 mL 50.0 mg/L的豆甾醇標(biāo)準(zhǔn)溶液,在室溫下吸附5~180 min,外加磁場分離,用0.45 μm的微孔薄膜過濾,收集上清液,以無水乙醇作參比,用紫外可見光譜儀測定豆甾醇質(zhì)量濃度。
1.3.2? 靜態(tài)吸附過程
取20 mg 3D-MMIPs或3D-MNIPs分別置于吸附管中,分別加入10 mL 10~50 mg/L 豆甾醇溶液,在室溫下吸附2 h,外加磁場分離,取上清液用紫外可見光譜儀測定豆甾醇質(zhì)量濃度。
1.3.2? 選擇性研究
取10.0 mg 3D-MMIPs加入2.0 mL質(zhì)量濃度均為25 mg/L的豆甾醇、膽固醇和β-谷甾醇混合溶液中吸附30 min,取上清液,用高效液相色譜測定豆甾醇、膽固醇和β-谷甾醇的質(zhì)量濃度。
2? ? 結(jié)果與討論
2.1? 三維印跡聚合物制備
3D-MMIPs的制備過程如圖1所示。先對多壁碳納米管羧酸化得到MWNTs-COOH;再采用水熱還原法在GO和MWNTs復(fù)合材料表面接枝Fe3O4納米顆粒,使GO和MWNTs復(fù)合材料具有磁性能。在三羥甲基氨基甲烷緩沖液中,加入上述三維磁性復(fù)合材料(3D-M-composite),以多巴胺為功能單體,豆甾醇為模板分子,通過多巴胺在弱堿性環(huán)境中的自聚合反應(yīng),接著用水洗出未反應(yīng)的多巴胺,再用乙酸/甲醇(2∶8,v/v)混合溶液將模板分子豆甾醇從印跡聚合物中洗脫出來,最后再用蒸餾水洗滌3次。這樣印跡聚合物中就形成對豆甾醇具有特異性識別的印跡位點(diǎn),成功地制備出新型三維豆甾醇磁性印跡復(fù)合材料(3D-MMIPs)。
2.2? 聚合物表征
2.2.1? 形貌分析
采用掃描電子顯微鏡對GO(A)、MWNTs(B)、3D-M-composite(C)、3D-MMIPs(D)的形貌進(jìn)行了表征,結(jié)果如圖2所示。由圖2可知,GO(A)為單層片狀結(jié)構(gòu);MWNTs(B)為管狀結(jié)構(gòu),并且相互纏繞,雜亂地堆砌在一起,碳納米管的粒徑大約為30 nm,3D-M-composite(C)為碳納米管和石墨烯的磁性雜化材料,在其表面能明顯觀察到磁性的Fe3O4微球。與圖3D-M-composite(C)相比,3D-MMIPs(D)中碳納米管的管徑明顯增大,約為68 nm,證明成功地合成了新型三維豆甾醇磁性印跡聚合物,由此可以估算出印跡層的厚度約為19 nm。
2.2.2? 紅外光譜分析
圖3中譜線MWNTs(b)和3D-M-composite(c)中618 cm-1處為Fe-O的收縮振動峰,這表明Fe3O4納米粒子成功地接枝在石墨烯和碳納米管復(fù)合材料表面;3D-MMIPs(d)中1 444 cm-1處為飽和碳?xì)滏I的彎曲振動峰,970 cm-1處反式取代烯烴,這表明豆甾醇成功修飾到3D-M-composite上。
2.3? 聚合物吸附性能研究
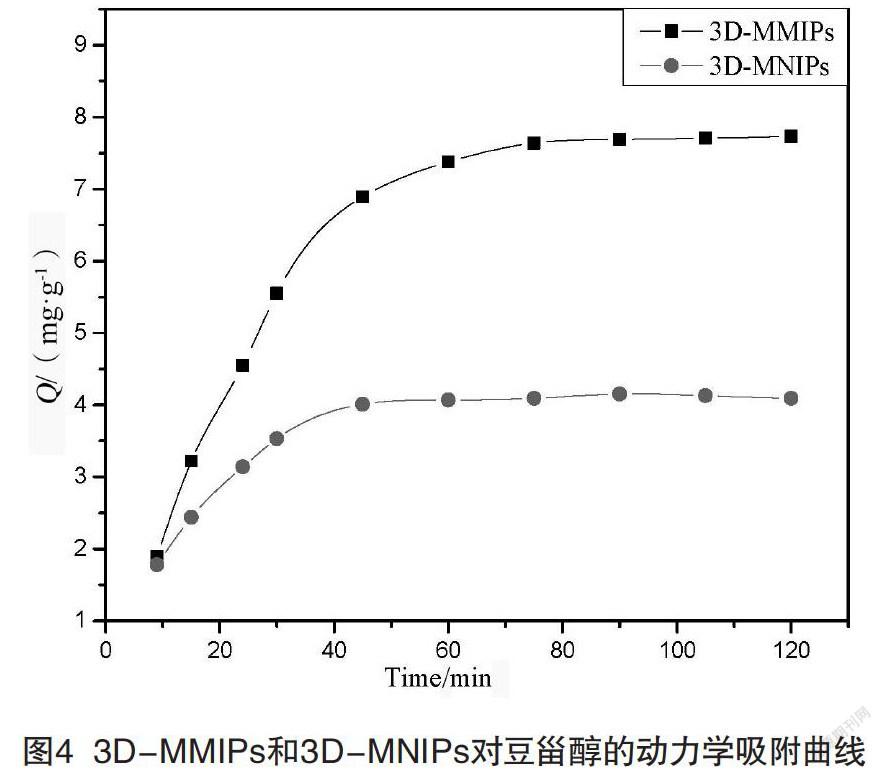
2.3.1? 動態(tài)吸附
圖4是3D-MMIPs對豆甾醇吸附容量對時間的關(guān)系圖。如圖4所示,吸附開始時,3D-MMIPs表面存在大量的結(jié)合位點(diǎn),使它能夠快速、良好地與豆甾醇結(jié)合。在吸附40 min前3D-MMIPs對豆甾醇的吸附速率增加很快。隨著時間的增加,豆甾醇結(jié)合了絕大多數(shù)的識別位點(diǎn),因此,吸附速率慢慢降低,直到60 min的時候,基本不再增加。這表明當(dāng)時間達(dá)到60 min時,3D-MMIPs的印跡孔穴對豆甾醇的吸附達(dá)到飽和。
2.3.2? 靜態(tài)吸附
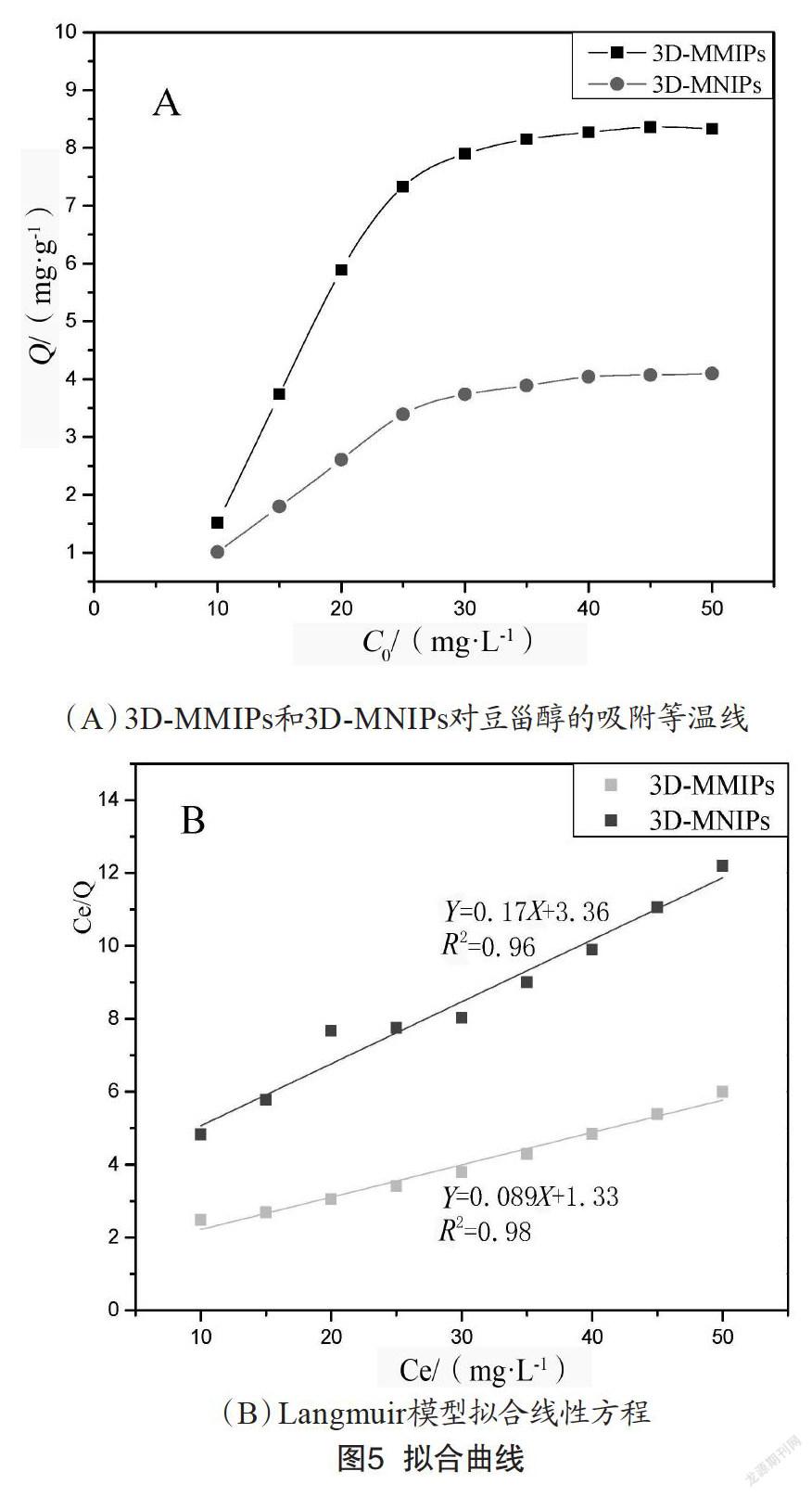
為了對該印跡材料的吸附容量進(jìn)一步研究,采用靜態(tài)平衡吸附實(shí)驗(yàn)探討3D-MMIPs和3D-MNIPs對豆甾醇的吸附等溫線,吸附容量由如下公式求得:
式中,Q表示聚合物的吸附量,mg/g;C0和C分別為溶液吸附前豆甾醇的濃度和吸附飽和后豆甾醇的質(zhì)量濃度,mg/L;V表示溶液的體積,mL;W表示聚合物的質(zhì)量,mg。由圖5(A)可知,3D-MMIPs對目標(biāo)分子豆甾醇的吸附性能明顯優(yōu)于3D-MNIPs對豆甾醇的吸附,主要是由于印跡聚合物殼層內(nèi)含有對模板分子具有特異吸附的結(jié)合位點(diǎn),通過官能團(tuán)與模板分子之間的氫鍵作用結(jié)合,增大了吸附容量。
采用Langmuir等溫吸附模型對3D-MMIPs的吸附進(jìn)一步擬合,結(jié)果如圖5(B)所示。Langmuir等溫吸附方程如下:
其中Q(mg/g)為豆甾醇的平衡吸附量,Qmax(mg/g)是等溫吸附方程的飽和吸附量,Ce(mg/L)是吸附平衡時豆甾醇質(zhì)量濃度,K(L/mg)為吸附平衡常數(shù)。以Ce/Q為Y軸,Ce為X軸作圖,擬合曲線圖,可以計算出K=0.026 L/mg,Qmax=11.24 mg/g。
2.3.3? 選擇性能研究
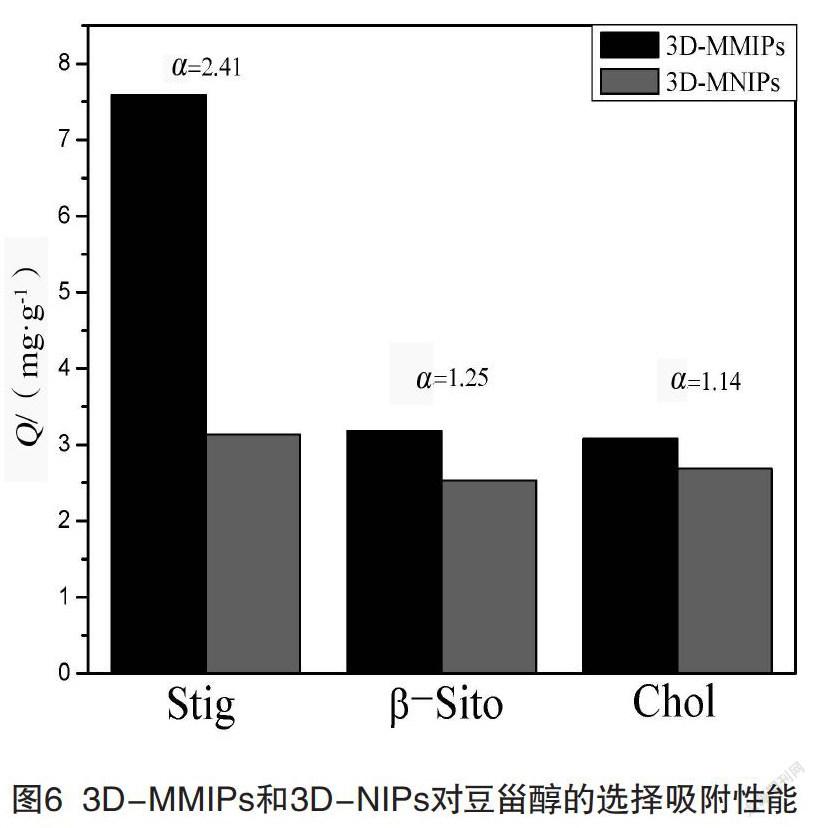
為考察3D-MMIPs和3D-MNIPs對豆甾醇的選擇吸附性能,選擇與豆甾醇結(jié)構(gòu)相似的膽固醇和β-谷甾醇作為競爭分子。結(jié)果如圖6所示,3D-MMIPs對豆甾醇的吸附量為7.59 mg/g,高于對膽固醇(3.18 mg/g)和β-谷甾醇(3.08 mg/g)的吸附量。3D-MMIP對豆甾醇、膽固醇和β-谷甾醇的印跡因子分別為2.41、1.25和1.14,3D-MMIP對膽固醇和β-谷甾醇的選擇性因子分別為1.93和2.11。這表明3D-MMIP表面有很多對豆甾醇具有特異性結(jié)合的位點(diǎn),可以與豆甾醇的結(jié)構(gòu)互補(bǔ),實(shí)現(xiàn)選擇性吸附。
3? ? 結(jié)語
采用多壁碳納米管和氧化石墨烯的復(fù)合材料作為載體,將Fe3O4磁性粒子修飾到復(fù)合材料表面;再利用水熱法對磁性復(fù)合材料進(jìn)行包覆。通過掃描電鏡、傅立葉變換紅外光譜等方法對該印跡復(fù)合材料進(jìn)行了表征,結(jié)果證明印跡層成功地包覆在三維磁性復(fù)合材料表面。吸附性能實(shí)驗(yàn)研究表明該三維磁性印跡材料對豆甾醇具有高選擇性和高吸附容量,其中飽和吸附容量為11.24 mg/g。
[參考文獻(xiàn)]
[1]胡? ?品,熊振湖,劉建明.分子印跡β-環(huán)糊精聚合物選擇性識別水中的雙酚A[J].高等學(xué)校化學(xué)學(xué)報,2012,33(9):2 111-2 116.
[2]饒? ?維,蔡? ?蓉,陳? ?星,等.氧化石墨烯表面新型磁性四溴雙酚A印跡復(fù)合材料的制備及吸附性能[J].高等學(xué)校化學(xué)學(xué)報,2013,34(6):1 353-1 359.
[3]閆? ?亮.三維碳基磁性印跡萃取材料制備及其性能研究[D].吉首:吉首大學(xué),2017.
[4]王? ?璇,張程琨,何金興.分子印跡固相微萃取纖維材料的制備研究進(jìn)展[J].化工新型材料,2018,46(6):235-238.
