微乳毛細(xì)管電動色譜-場放大富集法測定9種核苷類化合物
2019-08-30 08:35:28于曉章梁美娜李寧杰聶謹(jǐn)芳黃麗麗
分析科學(xué)學(xué)報 2019年4期
關(guān)鍵詞:分析
張 慶,于曉章,張 琳,梁美娜,李寧杰,聶謹(jǐn)芳,黃麗麗
(1.桂林理工大學(xué)環(huán)境科學(xué)與工程學(xué)院,廣西桂林541006;2.廣西環(huán)境污染控制理論與技術(shù)實(shí)驗(yàn)室,廣西桂林541006;3.福州大學(xué)食品安全與藥物化學(xué)教育部重點(diǎn)實(shí)驗(yàn)室,福建福州361006;4.桂林理工大學(xué)化學(xué)與生物工程學(xué)院,廣西桂林541006)
在基因工程、免疫學(xué)等現(xiàn)代生物技術(shù)的推動下,核苷及其衍生物等得到了廣泛的使用。尤其是核苷類化合物,其在抗病毒、抗腫瘤方面發(fā)揮了越來越重要的作用。目前,經(jīng)美國食品藥品監(jiān)督管理局(FDA)批準(zhǔn)的抗艾滋病藥物大多是核苷類衍生物,因此核苷類藥物有望成為新一代具有重要作用的藥物。目前,核苷類化合物的分析檢測方法主要有高效液相色譜法、薄層色譜法、柱色譜法及毛細(xì)管電色譜法等[1-5]。高效液相色譜法不僅分析時間長,而且流動相消耗大量的有機(jī)溶劑;薄層色譜法僅適用于少量樣品的分析;柱色譜法常需要加壓操作,不利于大規(guī)模樣品的測定;毛細(xì)管電色譜法具有快速、高效的特點(diǎn),但存在重現(xiàn)性較差,運(yùn)行時易產(chǎn)生氣泡等難題。因此,亟需探索和發(fā)展一種更為高效的分析介質(zhì)和更為靈敏的檢測手段,才能更進(jìn)一步發(fā)掘核苷類藥物在生物醫(yī)學(xué)中的作用。
微乳液毛細(xì)管電動色譜(MEEKC)是以微乳液作為分離介質(zhì),根據(jù)物質(zhì)的疏水性以及電遷移率的差異來實(shí)現(xiàn)分離的一種毛細(xì)管微分析技術(shù),該技術(shù)可以同時對電荷型/中性物質(zhì)、水溶性/脂溶性物質(zhì)進(jìn)行檢測[6-8]。MEEKC法具有高效、快速及分析對象廣等優(yōu)點(diǎn),但由于其檢測器多為紫外檢測器(UV),受到進(jìn)樣量和光程等因素的影響,靈敏度低,因此限制了MEEKC法在痕量分析中的應(yīng)用。為了提高該分析方法的檢測靈敏度和擴(kuò)大其應(yīng)用范圍,一些在線富集技術(shù),比如場放大富集(FASI)、大體積樣品堆積(LVSS)和離子選擇性耗盡掃集法等已被用于 MEEKC[9-10]。Zhang等[11]建立了一種 MEEKC-FASI分析尿液中嗎啡、可待因、納洛酮、海洛因、蒂巴因、可卡因、哌替啶、芬太尼、美沙酮9種麻醉劑的方法,該技術(shù)檢出限低至0.3μg/L。Chen等[12]首次建立了一種基于毛細(xì)管內(nèi)衍生化的膠束電動色譜法(MEKC)測定奶粉、液態(tài)奶、奶飲料和豆奶粉樣品中羥脯氨酸的方法。在最佳條件下,該方法可在7min內(nèi)完成衍生分離,脯氨酸的檢出限為1.6ng/mL。
本研究建立了一種新的MEEKC-FASI分析方法,對影響MEEKC分離效果和FASI富集效果等因素進(jìn)行了優(yōu)化,實(shí)現(xiàn)了腺嘌呤、鳥嘌呤、甲基腺苷、N6-甲基腺苷、胞苷、鳥苷、次黃嘌呤、巰嘌呤、氟尿嘧啶9種核苷類化合物的同時在線分析檢測。為了驗(yàn)證所建立的方法的可行性,進(jìn)行了尿樣和血清樣品的加標(biāo)回收率實(shí)驗(yàn),回收率范圍85.2%~113.0%,方法檢出限低至0.22μg/mL。
1 實(shí)驗(yàn)部分
1.1 主要儀器及試劑
安捷倫 HP3D毛細(xì)管電泳儀(Agilent technologies Inc),配備二極管陣列檢測器;KQ-100型超聲清洗儀(昆山超聲儀器有限公司);pHS-3C型精密酸度計(上海大普儀器有限公司);R200D分析天平(Sartorius);800型離心沉淀器(上海手術(shù)器械廠)。未涂層熔融石英毛細(xì)管(63cm,有效長度54.5cm,50μm i.d.×375μm o.d.)。
腺嘌呤(Aderine)、鳥嘌呤(Guanine)、甲基腺苷(Methladenosine)、N6-甲基腺苷(N6-methyladenosine)、胞苷(Cytidine)、鳥苷(Vernine)、次黃嘌呤(Hypoxanthine)、巰嘌呤(Mercaptopurine)、氟尿嘧啶(Fluorouracil)標(biāo)準(zhǔn)品(中國藥品生物制品鑒定所);十二烷基硫酸鈉(SDS)(Alfa Aesar);色譜純甲醇、乙腈(國藥集團(tuán)化學(xué)試劑有限公司);實(shí)驗(yàn)用水均為Milli-Q超純水。
血清和尿液(南京森貝伽生物科技有限公司)。
1.2 標(biāo)準(zhǔn)溶液和微乳液的配制
標(biāo)準(zhǔn)溶液:分別稱取適量的腺嘌呤、鳥嘌呤、甲基腺苷、N6-甲基腺苷、胞苷、鳥苷、次黃嘌呤、巰嘌呤、氟尿嘧啶標(biāo)準(zhǔn)品,用超純水溶解,配制成1 000μg/mL的標(biāo)準(zhǔn)儲備溶液,避光保存于4℃冰箱。使用時再稀釋至所需的濃度。微乳液:分別取13.5mg的SDS,11.0μL的正丁醇,8.0μL的乙酸乙酯和1470.0μL 10mmol/L的Na2B4O7緩沖溶液,置于2mL離心管中,超聲30min即可得到光學(xué)透明穩(wěn)定的微乳緩沖體系。固定微乳組成為質(zhì)量分?jǐn)?shù)為0.9%SDS、0.6%的正丁醇、0.5%的乙酸乙酯和98.0%10mmol/L的Na2B4O7緩沖溶液。
1.3 樣品制備
尿樣:取適量濃度的9種核苷類化合物標(biāo)準(zhǔn)溶液,混勻,加入尿液中,經(jīng)4 000r/min離心10min后取上層清液,用0.22μm濾膜過濾,再用10mmol/L Na2B4O7溶液稀釋10倍,制得尿樣。儲存在4℃冰箱備用。血清樣品:取適量濃度的9種核苷類化合物標(biāo)準(zhǔn)溶液混勻,加入血清中,經(jīng)離心處理10min(4 000r/min)后,取上層清液用0.22μm濾膜過濾,再用10mmol/L Na2B4O7溶液稀釋20倍,制得血清樣品。儲存在4℃冰箱備用。
1.4 樣品分析
毛細(xì)管預(yù)處理:依次用超純水、0.1mol/L HCl、超純水、0.1mol/L NaOH、超純水各沖洗30min,最后在電泳儀上用運(yùn)行緩沖液沖洗30min。待毛細(xì)管電泳(CE)基線和電流穩(wěn)定后,方可進(jìn)行CE操作分析。CE儀器操作:毛細(xì)管每次使用前依次用超純水、0.1mol/L NaOH溶液、超純水和緩沖液各沖洗10min;兩次運(yùn)行之間,依次用超純水和運(yùn)行緩沖液沖洗5min;每運(yùn)行5次,均需及時更換運(yùn)行緩沖液;每次更換運(yùn)行緩沖液后,均需用緩沖液平衡毛細(xì)管20min。所有溶液進(jìn)入CE儀器運(yùn)行前,均需經(jīng)0.22μm濾膜過濾,并超聲2min脫氣。安捷倫HP3D毛細(xì)管電泳儀檢測波長設(shè)置為200nm,柱溫保持為25℃。
2 結(jié)果與討論
2.1 MEEKC分離條件的優(yōu)化
2.1.1 緩沖溶液pH的選擇 緩沖溶液的pH值對 MEEKC的分離效果有顯著的影響[10-12]。考察了Na2B4O7緩沖溶液的pH在8.0~9.5之間變化時對9種核苷類化合物的分離情況。結(jié)果表明隨著pH值增大,分析時間逐漸延長。如圖1所示,當(dāng)pH為8.0和8.5時,9種核苷類化合物不能實(shí)現(xiàn)基線分離;當(dāng)pH為9.5時,N6-甲基腺苷(4號峰)和胞苷(5號峰)不能基線分離。綜合考慮分離度和遷移時間,選擇緩沖溶液的最佳pH值為9.0。
2.1.2 緩沖溶液濃度的選擇 使用低濃度(5~10mmol/L)的硼砂鹽或磷酸鹽能夠獲得較小的電流和較大的電滲流,因此上述2種緩沖溶液常被應(yīng)用于MEEKC體系中[13-14]。固定緩沖溶液的pH值為9.0,考察濃度5~15mmol/L Na2B4O7緩沖溶液對9種核苷類化合物分離效果的影響。如圖2所示,隨著硼酸鹽濃度的增加,分離度逐漸增大,被分析物的遷移時間也隨之延長,并且在15mmol/L的時候系統(tǒng)產(chǎn)生的焦耳熱變大,導(dǎo)致體系的穩(wěn)定變差。綜合考慮,緩沖溶液濃度選擇10mmol/L。

圖1 Na2B4O7緩沖溶液pH值對9種核苷類化合物分離效果的影響Fig.1 Effect of Na2B4O7buffer pH on the separation of 9nucleosidesseparation voltage:15kV;temperature:25 ℃;detection wavelength:200nm;electrokinetic injection:10kV×6s;microemulsion:0.9%SDS,0.6%1-butanol,0.5%ethyl acetate and 98.0%10mmol/L Na2B4O7buffer solution.1.a(chǎn)derine(2μg/mL);2.guanine(2 μg/mL);3.methladenosine (2.5 μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).

圖2 NaB4O7緩沖溶液濃度對9種核苷類化合物分離效果的影響Fig.2 Effect of NaB4O7buffer concentration on the separation of 9nucleosidesborate concentration:5,10and 15mmol/L;Other conditions were the same as Fig.1.1.a(chǎn)derine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
2.1.3 表面活性劑的選擇 SDS是MEEKC法中最常用的陰離子表面活性劑。實(shí)驗(yàn)研究了濃度為5~20mmol/L SDS對核苷類化合物分離效果的影響[11,15]。結(jié)果顯示,隨著SDS濃度的增大,由于電滲流的減小以及油滴表面負(fù)電荷的增多,遷移時間相應(yīng)的延長。當(dāng)SDS的濃度為15mmol/L時,次黃嘌呤(7號峰)和巰嘌呤(8號峰)遷移時間比較接近,分離度較差;當(dāng)SDS的濃度為20mmol/L時,雖然基線分離較好,但遷移時間較長;當(dāng)SDS的濃度較低時(5mmol/L),9種分析物分離度較差,腺嘌呤(1號峰)和鳥嘌呤(2號峰)甚至未基線分離。考慮微乳體系需要足夠的表面活性劑來維持穩(wěn)定,最終選擇10mmol/L的SDS為最佳濃度。
2.1.4 助表面活性劑的選擇 正丁醇是MEEKC法中最常用的助表面活性劑[11,16-17]。微乳體系中加入一定濃度的正丁醇,可以增加微乳滴液膜的機(jī)械強(qiáng)度,有利于維持MEEKC運(yùn)行時微乳液的穩(wěn)定。但如果正丁醇過量,多余的正丁醇分子會與表面活性劑(如SDS)的極性基團(tuán)締合,導(dǎo)致微乳液膜松散,降低微乳體系的穩(wěn)定性,容易出現(xiàn)破乳現(xiàn)象[9]。因此,考察了正丁醇含量對9種核苷類化合物分析效果和微乳液穩(wěn)定性的影響。結(jié)果顯示,正丁醇質(zhì)量分?jǐn)?shù)由0.3%増至0.9%時,隨著正丁醇含量的增加,遷移時間隨之延長;但正丁醇質(zhì)量分?jǐn)?shù)增到1.2%,遷移時間縮短,且分離度很差。綜合考慮各分析物的分離度和靈敏度,選擇0.6%的正丁醇為最佳助表面活性劑。
2.1.5 油相種類及濃度的選擇 油相一般采用正己烷、正庚烷、正辛烷、乙酸乙酯等化合物[11,18]。鑒于乙酸乙酯與水之間的表面張力更小,因此將其作為微乳體系的油相微乳液,僅需少量的表面活性劑即可維持體系的穩(wěn)定。以乙酸乙酯為油相,考察了其濃度對9種核苷類化合物分離效果的影響。在0.25%~0.75%(質(zhì)量分?jǐn)?shù))乙酸乙酯含量范圍內(nèi),9種分析物均完全分離,且選擇性和靈敏度均無明顯的變化。為確保微乳體系能在較低的SDS濃度下維持穩(wěn)定,故確定0.5%乙酸乙酯為油相。
2.1.6 分離電壓的選擇 分離電壓不僅影響分離效率,也是影響分析時間的一個重要參數(shù)[11,18-24]。研究了在10、15、20、25kV的條件下,分離電壓對9種核苷類化合物分離效果和靈敏度的影響。結(jié)果發(fā)現(xiàn),隨著分離電壓的增大,9種分析物的遷移時間逐漸縮短。當(dāng)電壓為10kV時,分析物的分離時間較長,且?guī)€嘌呤(8號峰)和氟尿嘧啶(9號峰)響應(yīng)值都有所下降;當(dāng)電壓為20kV和25kV時,分析物的分離度均有所下降,原因可能為體系的電流較大導(dǎo)致毛細(xì)管焦耳熱較大,引起電流和基線不穩(wěn)定。綜合考慮,實(shí)驗(yàn)選擇15kV為最合適的分離電壓。綜上所述,確定最佳的實(shí)驗(yàn)條件為:微乳液組成為質(zhì)量分?jǐn)?shù)0.9%的SDS、0.6%的正丁醇、0.5%的乙酸乙酯和98.0%的10mmol/L Na2B4O7緩沖液(pH=9.0),分離電壓為15kV。9種核苷類化合物在最佳微乳組成條件下的MEEKC譜圖如圖3所示。從圖中可以看出,除了N6-甲基腺苷(4號峰)和胞苷(5號峰)分離度相對較小外,9種分析物在12min內(nèi)基本實(shí)現(xiàn)基線分離。
2.2 富集條件的優(yōu)化
2.2.1 樣品稀釋劑的選擇 FASI主要基于高壓電場下樣品溶液和背景緩沖溶液電導(dǎo)的差異而實(shí)現(xiàn)樣品富集的,因此樣品基體對FASI富集效果有重要的影響[7,23-24]。分別以10mmol/L Na2B4O7緩沖溶液、微乳液、0.1mmol/L NaOH溶液、甲醇和水為樣品稀釋劑,考察了樣品稀釋劑對9種核苷類化合物靈敏度和分離度的影響。結(jié)果發(fā)現(xiàn),當(dāng)樣品稀釋劑為微乳液時,雖然鳥嘌呤的響應(yīng)值明顯得到提高,但其余分析物的分離度下降,氟尿嘧啶甚至不出峰;0.1mmol/L NaOH溶液作為樣品稀釋劑時,腺嘌呤響應(yīng)值下降,N6-甲基腺苷峰變寬;甲醇為樣品稀釋劑時,鳥嘌呤、巰嘌呤和氟尿嘧啶響應(yīng)值下降;水為樣品稀釋劑對各分析物的靈敏度顯著下降。在10mmol/L Na2B4O7緩沖溶液中,9種核苷類化合物的靈敏度均得到提高,且具有良好的分離度。綜合考慮,選擇10mmol/L Na2B4O7緩沖溶液作為樣品稀釋劑。
2.2.2 進(jìn)樣條件的選擇 場放大進(jìn)樣技術(shù)的進(jìn)樣量主要取決于進(jìn)樣電壓和進(jìn)樣時間。考察進(jìn)樣電壓在14~24kV范圍內(nèi)9種核苷類化合物的靈敏度變化情況。隨著進(jìn)樣電壓的增大,檢測靈敏度也隨之提高;當(dāng)進(jìn)樣電壓超過22kV時,各分析物的峰形展寬,分離度開始下降,說明此時樣品富集失敗。原因可能為:(1)FASI富集過程中,進(jìn)樣量隨進(jìn)樣電壓(或者進(jìn)樣時間)的增大而增大,當(dāng)進(jìn)樣量超過毛細(xì)管所能承受的范圍時(FASI進(jìn)樣量只能低于毛細(xì)管總體積的10%),由于樣品擴(kuò)散作用,導(dǎo)致峰形展寬,靈敏度和分離度下降;(2)高進(jìn)樣電壓使樣品區(qū)帶產(chǎn)生大量的焦耳熱,引起毛細(xì)管內(nèi)溫度驟升,影響體系電流和基線的穩(wěn)定;(3)樣品稀釋劑和背景緩沖溶液存在電導(dǎo)差異,過高的進(jìn)樣電壓,可能引起體系產(chǎn)生氣泡,導(dǎo)致富集失敗。進(jìn)樣電壓為22kV,考察進(jìn)樣時間對峰高的影響。進(jìn)樣時間從5s增加到15s的過程中,被分析物的峰高僅在5~10s的范圍內(nèi)是隨著進(jìn)樣時間的增加而增高。當(dāng)進(jìn)樣時間為15s時,峰形展寬,分離效率下降,故以10s(22kV)為最佳進(jìn)樣時間。
2.2.3 FASI與常規(guī)進(jìn)樣對比及富集倍數(shù) 文獻(xiàn)報道[11,25]在樣品電動進(jìn)樣前壓力注入一段水塞可以提高富集效果。實(shí)驗(yàn)結(jié)果表明,當(dāng)以3kPa壓力進(jìn)6s水柱時,富集效果最好。在最佳的MEEKC條件下,對比了FASI和常規(guī)進(jìn)樣兩種方式對9種核苷類化合物分離選擇性和檢測靈敏度的影響。如圖4所示,在不影響核苷類化合物分離度和遷移時間的情況下,采用FASI進(jìn)樣方式可顯著提高各分析物的檢測靈敏度。
對比常規(guī)進(jìn)樣方式,場放大富集的倍數(shù)可表示為:f=(h/h0)×(c0/c)。其中,h0和h分別為常規(guī)進(jìn)樣和采用FASI進(jìn)樣后溶質(zhì)的峰高,c0和c分別為兩種情況下溶質(zhì)的濃度。依據(jù)公式可以計算出9種核苷類化合物的富集倍數(shù)分別為:腺嘌呤(33倍)、鳥嘌呤(12倍)、甲基腺苷(38倍)、N6-甲基腺苷(14倍)、胞苷(18倍)、鳥苷(20倍)、次黃嘌呤(21倍)、巰嘌呤(8.8倍)及氟尿密度(14倍)。

圖3 最佳MEEKC條件下9種核苷類化合物的電泳譜圖Fig.3 Electropherogram of 9nucleosides under the optimized MEEKC conditionsseparation voltage:15kV;temperature:25 ℃;detection wavelength:200nm;electrokinetic injection:10kV×6s;microemulsion solution:0.9%SDS,0.6%1-butanol,0.5%ethyl acetate and 98.0%10mmol/L Na2B4O7buffer solution.1.a(chǎn)derine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
2.3 線性范圍和檢出限
配制一系列不同濃度的9種核苷類化合物的混合標(biāo)準(zhǔn)溶液,在最佳富集條件下進(jìn)行MEEKC測定。各分析物的線性范圍、線性回歸方程、相關(guān)系數(shù)及檢出限(S/N=3)如表1所示。9種核苷類化合物在線性范圍內(nèi)相關(guān)系數(shù)大于0.9960,相對標(biāo)準(zhǔn)偏差(RSD)均小于5.74%。
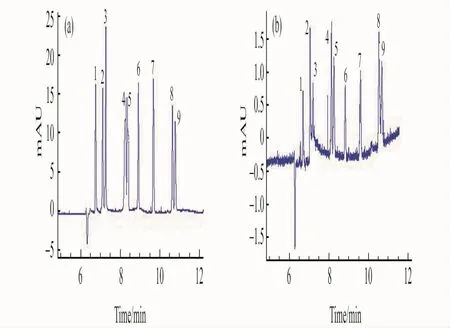
圖4 MEEKC-FASI(a)與常規(guī)MEEKC(b)進(jìn)樣的9種核苷類化合物毛細(xì)管電泳譜圖Fig.4 Elecropherograms of 9nucleosides for MEEKC-FASI(a)and conventional MEEKC (b)(a):MEEKC-FASI:water plug(30mbar,6s);electrokinnetic injection(22kV×10s).1.a(chǎn)derine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL);(b):normal electrokinetic injection,10kv×5s;1.a(chǎn)derine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
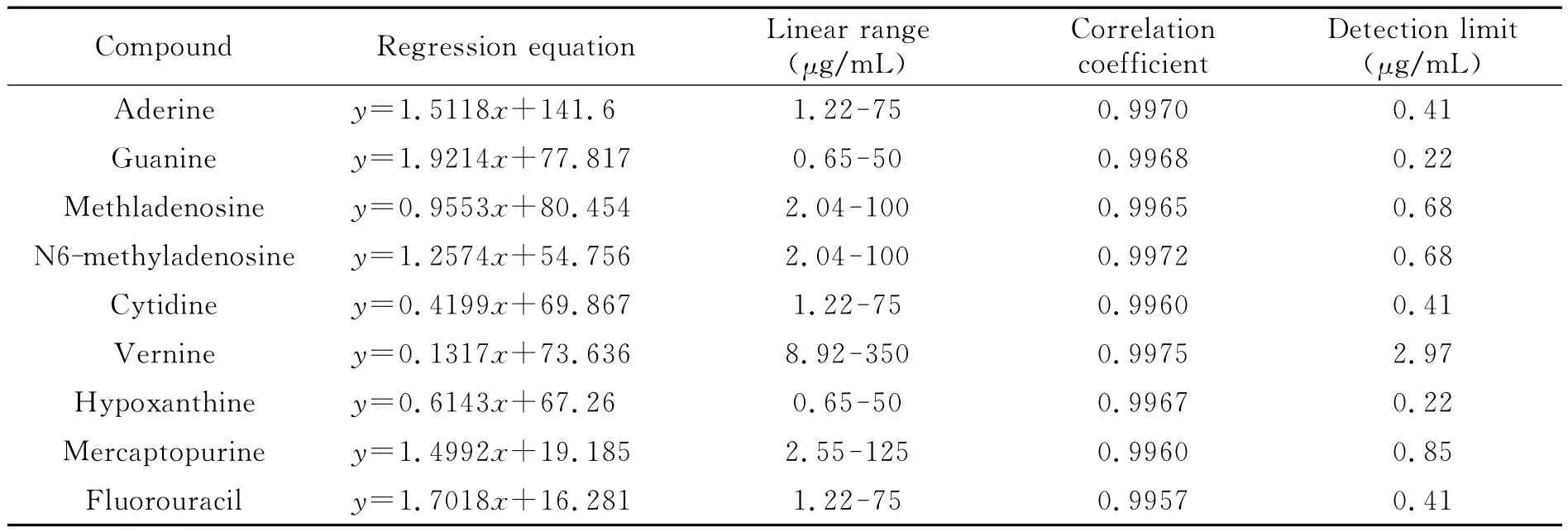
表1 9種核苷類化合物的線性回歸方程、線性范圍、相關(guān)系數(shù)和檢出限Table 1 Regression equations,linear ranges,correlation coefficients and detection limits for 9nucleosides
2.4 回收率和精密度
2.4.1 尿樣的測定 根據(jù)1.3節(jié)方法制備尿樣,按照1.4節(jié)所述的條件進(jìn)行分析,尿樣的電泳譜圖如圖5所示。由圖5可以看出,除了N6-甲基腺苷(4號峰)和胞苷(5號峰)峰形有略微展寬,分離度變小,以及巰嘌呤(8號峰)和氟尿密度(9號峰)的分離度有所下降以外,其他分析物均沒有受到基質(zhì)的影響。分別向尿樣中加入2個不同濃度水平的9種核苷類化合物的標(biāo)準(zhǔn)混合溶液進(jìn)行加標(biāo)回收率實(shí)驗(yàn)。每個濃度平行測定3次,計算相對標(biāo)準(zhǔn)偏差(RSD)。如表2所示,9種核苷類化合物的回收率介于91.2%~113.0%之間,RSD均小于5.9%。加標(biāo)回收實(shí)驗(yàn)結(jié)果表明,所建立的方法穩(wěn)定可靠。

圖5 9種核苷類化合物尿樣(a)和空白(b)的電泳譜圖Fig.5 Elecropherograms of 9nucleosides in urine(a)and in blank samples(b)experimental conditions same as the Fig.4(a).1.a(chǎn)derine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
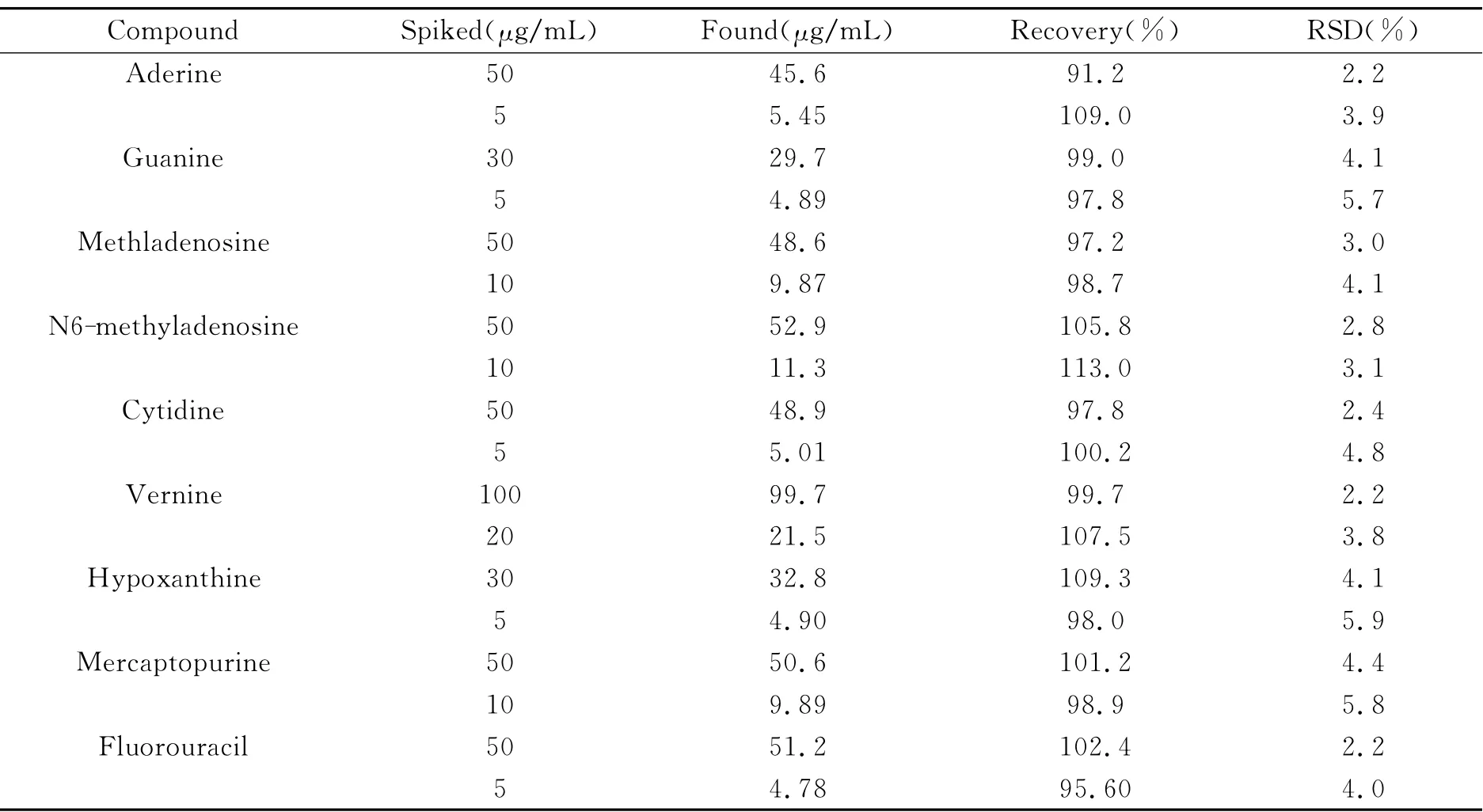
表2 尿樣中9種核苷類化合物的加標(biāo)回收率(n=3)Table 2 Recoveries of 9nucleosides spiked in urine samples(n=3)
2.4.2 血清樣品的測定 血清樣品根據(jù)1.3節(jié)步驟處理,按照1.4節(jié)所述的條件進(jìn)行分析。同時為驗(yàn)證所建立的方法應(yīng)用于血清樣品中9種核苷類化合物檢測結(jié)果的可靠性,進(jìn)行了2個濃度水平的加標(biāo)回收率實(shí)驗(yàn)。每個濃度平行測定3次,計算相對標(biāo)準(zhǔn)偏差(RSD),得到9種核苷類化合物的回收率介于85.2%~111.8%之間,RSD均小于8.2%。
3 結(jié)論
本文建立了一種快速、靈敏的同時測定9種核苷類化合物的微乳液毛細(xì)管電動色譜-場放大富集分離分析方法,分別研究了MEEKC分離條件、FASI富集條件等因素的影響。在最佳的實(shí)驗(yàn)條件下,9種核苷類化合物可在12min內(nèi)實(shí)現(xiàn)基線分離,且被測物濃度與峰電流呈良好的線性關(guān)系,檢出限(S/N=3)低至0.22μg/mL。此外,將所建立的方法應(yīng)用到尿樣和血清樣品分析,成功地檢測到了9種核苷類化合物。該方法具有簡單、靈敏等特點(diǎn),有望在藥物分析等領(lǐng)域得到廣泛的應(yīng)用。
猜你喜歡
現(xiàn)代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機(jī)設(shè)計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業(yè)技術(shù)(2016年15期)2016-12-01 05:31:22
當(dāng)代經(jīng)濟(jì)研究(2016年5期)2016-12-01 03:12:05
現(xiàn)代農(nóng)業(yè)(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
財經(jīng)界(學(xué)術(shù)版)(2015年20期)2015-12-23 09:20:13
中國中醫(yī)藥現(xiàn)代遠(yuǎn)程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學(xué)學(xué)報(社會科學(xué)版)(2014年3期)2014-04-16 04:38:31
