超高效液相色譜-四極桿靜電場軌道離子阱質譜同時檢測 動物源性食品中20種β-受體阻滯劑及代謝物
2019-08-29 08:33:26聶雪梅董旭陽許秀麗
色譜 2019年9期
聶雪梅, 董旭陽,2, 許秀麗, 張 峰*
(1. 中國檢驗檢疫科學研究院, 北京,100176; 2. 陜西科技大學, 西安,710021)
β-受體阻滯劑(β-blockers, BBS)可選擇性地與β腎上腺素受體結合,拮抗兒茶酚胺和神經遞質對β受體的激動作用,在治療心血管和心功能不全等疾病中有非常重要的作用[1],但β-受體阻滯劑的不當使用會導致心肌耗氧量和血管阻力增加,氧自由基和心肌細胞凋亡[2,3]。在畜牧養殖業中,β-受體阻滯劑常被非法添加在飼料中,通過降低動物的運動量來提高飼料轉化率;在動物屠宰或運輸前數小時也會被非法注射到動物體內來緩解應激反應,降低動物在運輸壓力下的發病率和死亡率[4,5]。另外β-受體阻滯劑進入動物體內會進一步發生代謝,生成藥理活性更高的代謝物[6]。如卡維地洛的代謝物4-羥基苯基卡維地洛,其腎上腺素受體阻滯效能比原藥高約13倍,而該類藥物及其代謝物在動物體內殘留風險較高,可引起人類急性中毒的癥狀[7]。為滿足監管的要求,建立一種快速篩查β-受體阻滯劑及其代謝物殘留的分析方法是非常必要的。
目前,世界上一些地區和組織已經為動物源性食品中卡拉洛爾設定了最大殘留限量,以保護公眾健康。歐盟和國際食品法典委員會規定了食用動物組織中卡拉唑醇的最大殘留限量,豬腎中為25 μg/kg,牛腎中為15 μg/kg[8]。但對于β-受體阻滯劑的代謝物(如4-羥基苯基卡維地洛等),尚未設定殘留限量。我國目前既未批準BBS作為獸藥使用,也未明確BBS及其代謝物的相關限量要求。因此,建立動物源食品中β-受體阻滯劑及其代謝物的高通量篩查分析方法具有重大意義。
為了監控β-受體阻滯劑的非法使用,全世界的監管機構正在測試動物源性食品中的此類藥物。常用的分析方法有酶聯免疫吸附法(ELISA)、液相色譜法(LC)和液相色譜-質譜法(LC-MS)、液相色譜-熒光檢測法(LC/UV)、氣相色譜法(GC)、氣相色譜-質譜法(GC/MS)[9,10]等。其中ELISA定量不準確且操作程序復雜,檢測后需要其他方法驗證。液相色譜法和液相色譜-熒光檢測法的靈敏度較低,無法滿足對痕量化合物的分析要求。氣相色譜法和氣相色譜-質譜法具有較好的靈敏度,但前處理需衍生化,費時較長并增加了樣品污染的可能性。液相色譜-質譜法作為目前應用較多的方法,可以進行多殘留定性、定量檢測,方便快速[11,12]。近年來,超高效液相色譜-四極桿/靜電場軌道阱高分辨質譜(UHPLC-Q-Orbitrap MS)在動物源性食品中的農獸藥殘留檢測領域逐漸普及,但對于肉類食品中β-受體阻滯劑及其代謝物殘留分析的篩查分析方法的相關研究報道較少。本實驗采用Q-Orbitrap高分辨質譜分析系統,建立了同時測定動物源食品中20種β-受體阻滯劑類藥物及代謝產物的分析方法。該方法簡單,快速,靈敏度高,為動物源性食品的安全提供了保障。
1 實驗部分
1.1 材料與試劑
乙酸銨、氯化鈉、乙二胺四乙酸二鈉(分析純,北京化工廠);甲醇、乙腈(色譜純,美國Thermo Fisher公司);甲酸(純度>99%,北京百靈威科技有限公司)。
標準品:卡拉洛爾、馬來酸噻嗎洛爾、納多洛爾、吲哚洛爾、美托洛爾(德國Dr. Ehrenstorfer公司);醋丁洛爾、卡維地洛、噴布洛爾、普萘洛爾、鹽酸倍他洛爾、阿普洛爾、氧烯洛爾、塞利洛爾、富馬酸比索洛爾(法國EP公司)鹽酸拉貝洛爾純度(美國U.S.P公司);二醋洛爾、氯拉洛爾、艾司洛爾、7-羥基普萘洛爾、羥基噻嗎洛爾、羥苯基卡維地洛、羥基阿替洛爾(加拿大Toronto Research Chemicals公司);以上試劑純度均大于96%。
1.2 儀器與設備
Q-Exactive超高效液相色譜-四極桿/靜電場軌道阱高分辨質譜聯用儀(美國Thermo Fisher公司); Milli-R04純水儀(德國Millipore公司); Allegra X-22R高速冷凍離心機(美國Beckman Coulter公司); ML104/02分析天平(梅特勒-托利多上海有限公司); VORTEX GENIUS3渦旋混合器(德國IKA公司);全溫振蕩培養器(德國Sartorius公司)。
1.3 溶液的配制
準確稱取20種β-受體阻滯劑及其代謝物各1 mg(精確至0.000 1 g),分別置于20 mL的棕色小瓶中,各加入5.00 mL的甲醇,振蕩搖勻,待固體完全溶解后,再加入5.00 mL的甲醇,配制成100 mg/L的標準儲備液,在-20 ℃冰箱中儲存待用。分別精密移取上述20種β-受體阻滯劑及代謝物標準儲備液0.50 mL至10 mL棕色小瓶中,混勻,配制成5 mg/L的β-受體阻滯劑及代謝物的混合標準溶液,并放置于-20 ℃冰箱中保存待用。
稱取15.400 0 g乙酸銨,溶于水中,用乙酸調pH值為5.2。充分搖勻后,加入37.5 g乙二胺四乙酸(EDTA)二鈉并定容至1.0 L,配制成EDTA提取液并放置于4 ℃冰箱中保存待用。
1.4 樣品前處理
將肉類樣品攪碎后,稱取1.000 0 g樣品到50 mL聚丙烯離心管中,加入5.00 mL乙二胺四乙酸提取液和20.0 μLβ-葡糖苷酸酶/芳基硫酸酯酶后,在旋轉器上混合1 min, 50 ℃下全溫振蕩培養器中300 r/min振蕩酶解2 h。待其冷卻至室溫后,用3.0 mol/L氫氧化鈉溶液pH調節至10。將乙腈(20.00 mL)、氯化鈉(2.000 0 g)依次加入到樣品中,搖勻振蕩提取30 min后在4 ℃下以10 000 r/min離心15 min。取上清液過0.22 μm尼龍膜到進樣小瓶中,上機檢測。
1.5 儀器條件
1.5.1色譜條件
色譜柱:ALORICH Ascentis?C8(10 cm×4.6 mm, 3 μm)。流動相:0.1%(v/v)的甲酸水溶液(A)、乙腈(B)。梯度洗脫程序:0~0.5 min, 5%B; 0.5~9.0 min, 5%B~95%B; 9.0~12.5 min, 95%B; 12.5~14.0 min, 95%B~5%B; 14.0~15.0 min, 5%B。柱溫箱溫度為30 ℃,流速為0.3 mL/min,進樣體積為5 μL。
1.5.2質譜條件
Q-Orbitrap HRMS配備了加熱電噴霧電離源(HESI),掃描模式為正離子掃描,數據依賴模式(Full MS/dd-MS2),質量掃描范圍m/z100~500。一級分辨率70 000,二級分辨率35 000。歸一化碰撞能量(normalized collision energies, NCE)設定為15%、25%和35%,噴霧電壓設定為3.0 kV,毛細管溫度和輔助氣體加熱器溫度均為350 ℃。20種化合物的質譜參數見表1。
1.6 基質效應
在樣品中,除分析物以外的組分都被稱為基質。基質中存在的干擾物會與目標物共洗脫,改變待測組分的離子化效率,降低方法的靈敏度,影響分析結果的準確性[13,14]。基質效應(ME)根據相同濃度的分析物在純有機溶液中與在空白基質溶液中的響應比值來評估[15]:
ME=B/A×100%
(1)
其中A為純溶劑中待測物的響應值,B為樣品基質中添加的相同含量待測物的響應值。動物源性食品成分復雜,基質效應明顯。為了最大限度地消除基質效應,本實驗在空白基質中加入系列標準溶液建立校正曲線。這樣標準空白基質和待測化合物具有相同的離子化環境,從而有效消除基質效應。

表 1 20種目標化合物的色譜-質譜參數Table 1 LC-MS parameters of the 20 target compounds
2 結果與討論
2.1 色譜條件的優化
在對目標化合物進行檢測時,應最大程度考慮色譜峰峰形的對稱性,以確保開發的檢測方法以峰面積對目標物進行定量時,有更加接近真值的結果[16]。因此,色譜柱與流動相的選擇非常關鍵。
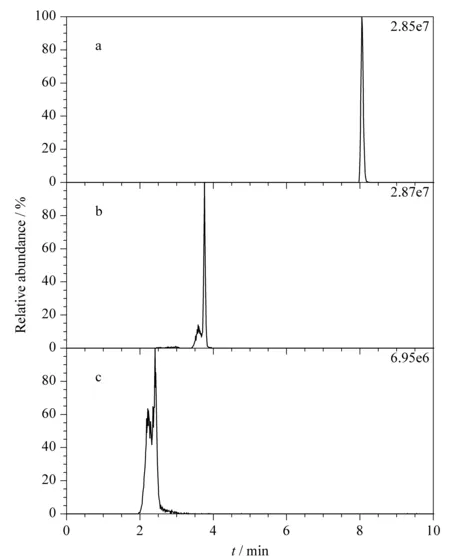
圖 1 3種色譜柱對卡拉洛爾的分離效果Fig. 1 Separation of carazolol on the three chromatographic columnsa. ALORICH Ascentis? C8 (10 cm×4.6 mm, 3 μm); b. Waters ACQUITY UPLC? BEH C18(50 mm×2.1 mm, 1.7 μm); c. Waters BEH HILIC (100 mm×2.1 mm, 1.7 μm).
本實驗考察了Waters ACQUITY UPLC?BEH C18(50 mm×2.1 mm, 1.7 μm)、Waters BEH HILIC (100 mm×2.1 mm, 1.7 μm)和ALORICH Ascentis?C8(10 cm×4.6 mm, 3 μm)這3種色譜柱對目標物的分離效果。色譜柱優化時,流動相采用實驗室常用流動相(甲醇和0.1%(v/v)甲酸水溶液)。以卡拉洛爾為例,3種色譜柱的分離效果如圖1所示。觀察實驗結果發現,HILIC色譜柱對目標物進行分離時,色譜峰分叉嚴重,這可能是由于β-受體阻滯劑類藥物極性范圍較寬,而適用于極性化合物的HILIC色譜柱對β-受體阻滯劑類化合物的分離效果不夠理想。C18色譜柱對目標物進行分離時,峰形較差,這對目標物定性及定量結果有很大的影響。而C8色譜柱得到的色譜峰峰形良好,適合用于β-受體阻滯劑類藥物的分離。該色譜柱填料為高純硅膠,與普通C18色譜柱相比,具有更好的親水性,對于β-受體阻滯劑色譜峰峰形的改善、峰寬減小等有很大幫助。
除色譜柱外,流動相的選擇對待測物的分離及質譜的檢測結果也有很大的影響。色譜柱選定后,通過實驗對比了甲醇、乙腈與0.1%(v/v)甲酸水溶液作為流動相時目標物的分離情況。實驗結果如圖2所示,乙腈作為有機相時目標物響應強度較甲醇高。由于本實驗采用正離子模式檢測,因此在水相中加入少量酸可增強目標物的響應。對比發現,乙腈-0.1%(v/v)甲酸水溶液作流動相時目標物的色譜峰峰形好,響應高。在優化的儀器條件下,20種β-受體阻滯劑類藥物及其代謝物的色譜圖見圖3。

圖 2 不同流動相對卡拉洛爾的分離效果Fig. 2 Separation effect of different mobile phases on carazolol a. methanol-0.1% (v/v) formic acid; b. acetonitrile-0.1% (v/v) formic acid.

圖 3 20種β-受體阻滯劑及代謝物的色譜圖Fig. 3 Chromatograms of the 20 β-blockers and metabolites
2.2 前處理條件的優化
動物源性食品基質復雜,并且本實驗要同時檢測20種β-受體阻滯劑及代謝物。因此,在前處理過程中進行凈化方法的選擇時,在保證目標化合物物回收率良好的前提下,需要最大程度除去雜質,以提高目標物的靈敏度。
2.2.1QuEChERS凈化
移取振蕩提取后的樣品上清液4 mL到10 mL離心管中,加入0.9 g無水硫酸鎂、150 mg C18和150 mg乙二胺-N-丙基硅烷,振蕩2 min后靜置,取上清液過0.22 μm濾膜。上機檢測后發現,加標樣品中卡拉洛爾、氧烯洛爾、普萘洛爾、阿普洛爾、比索洛爾、噻嗎洛爾等10種目標化合物的回收率均小于60%。

表 2 20種目標化合物的線性方程、相關系數、線性范圍、檢出限和定量限Table 2 Linear equations, correlation coefficients (r2), linear ranges, LODs and LOQs of the 20 target compounds
Y: peak area;X: mass concentration, μg/L.
2.2.2固相萃取
將振蕩提取后的樣品靜置分層后,移取2 mL上清液分別過親水親脂分配固相萃取柱(Waters Oasis HLB, 200 mg/6 mL)和混合強陽離子固相萃取柱(Waters Oasis MCX, 150 mg/6 mL)。HLB柱用甲醇-水(5∶95, v/v)和甲醇-乙腈(1∶9, v/v)依次淋洗。MCX柱用含有5%(v/v)氨水的甲醇淋洗。實驗結果發現,使用MCX柱進行凈化時,目標物的回收率更高。這是由于HLB柱是共價鍵結合分配機理,因此目標物回收率不夠理想。而MCX柱的原理為苯磺酸型強陽離子交換機理,是將磺酸基鍵合在高度交聯的聚(苯乙烯-二乙烯基苯) (PS/DVB)表面,具有反相和交換雙重保留性能,對堿性化合物有良好的保留性能[17,18]。但還是有少量化合物的回收效果不佳。
2.2.3高速低溫離心
將振蕩提取后的樣品于4 ℃下10 000 r/min離心15 min。取上清液過0.22 μm濾膜進樣。實驗結果發現,20種目標化合物的平均回收率在60.37%~100.84%,回收率良好。這可能是由于該方法減少了凈化步驟,避免了目標化合物在凈化過程中的損失。除此之外,該方法操作簡單,試劑使用量少。因此,本研究采用該方法對樣品進行凈化。
2.3 方法學考察
2.3.1線性范圍和檢出限
分別添加0.1、0.2、0.5、1、2、5、10 μg/L的目標化合物混合標準物質到空白基質溶液中。以20種目標化合物的響應峰面積-對應的質量濃度繪制工作曲線。以峰面積(Y)為縱坐標,質量濃度(X, μg/L)為橫坐標,繪制標準曲線,20種化合物在0.1~10 μg/L范圍內呈良好的線性關系,線性相關系數(r2)均大于0.99。在空白樣品中分別添加一系列濃度的目標化合物混合標準溶液,進行前處理和測定,以3倍信噪比確定檢出限(LOD),以10倍信噪比確定定量限(LOQ)。實驗結果表明,20種目標化合物的檢出限為1~5 μg/kg,定量限為2~10 μg/kg。具體結果見表2。
2.3.2回收率與精密度
空白樣品中添加低、中、高3個水平的20種目標化合物混合標準溶液,每個水平進行6次試驗。結果表明,20種化合物的平均回收率為60.37%~100.84%,相對標準偏差小于10%(見表3)。其準確度與精密度能滿足殘留檢測的要求,表明該分析方法可靠、準確,適合此類物質的定量分析。
2.4 實際樣品的分析
購買來自不同國家和地區的市售豬肉共20份,將本實驗建立的方法應用到20份樣品中β-受體阻滯劑及代謝物殘留的檢測中。
實驗結果表明,實際樣品中均未檢出該類物質,未發現陽性樣品。

表 3 20種目標化合物在3個添加水平下的平均回收率和相對標準偏差(n=6)Table 3 Average recoveries and relative standard deviations of the 20 target compounds at three spiked levels (n=6)
3 結論
本研究建立了超高效液相色譜-四極桿/靜電場軌道離子阱質譜快速篩查方法,實現了20種β-受體阻滯劑及代謝物的定性、定量分析。該法操作簡單,重現性好,分辨率高,能同時對動物源性食品中20種β-受體阻滯劑及代謝物殘留進行篩查定量。
