鋰離子電池正極材料Li2MnO3稀土摻雜的第一性原理研究*
2019-08-27 06:57:10鄭路敏鐘淑英徐波歐陽楚英
物理學報 2019年13期
關鍵詞:結構
鄭路敏 鐘淑英 徐波 歐陽楚英
(江西師范大學物理與通信電子學院,南昌 330022)
1 引 言
鋰離子電池自誕生以來給社會科技的發展做出了巨大的貢獻,特別是應用在各種電子設備上[1].隨著電子設備的更新換代,越來越多的電子設備要求更高容量、更快充放電速率以及更長循環壽命的鋰離子電池.其中,在電池正極材料的改進上,摻雜作為一種有效的策略,對電池的性能有很大的改善[2].適當的摻雜可以有效抑制電極材料在充放電過程中的結構變化,從而提高鋰離子電池的循環性能和倍率性能[3].
稀土元素具有大的離子半徑,高的電子電荷,以及強的自極化能力,作為摻雜元素,不僅對于尖晶石型鈷鐵氧體的電子結構和磁性有明顯的調節作用[4],而且在鋰離子電池正極材料的改性上起到了積極的作用.通過稀土摻雜的方式可以有效改善正極材料的性能.近些年,實驗方面對于稀土元素摻雜鋰離子電池正極材料的研究有很多,比如:LiCoO2[5?7],LiMn2O4[8?12],LiFePO4[13,14]和LiNi1/3Co1/3Mn1/3O2[15]等.Yang等[16]報道了LiMn2O4電池正極材料的稀土摻雜,摻雜后形成的LiMn2–xRExO4(x=0.01,RE=Y,Nd,Gd,Ce)體系晶格參數由于摻雜稀土離子而增大,形成更為穩定的骨架結構,顯著提高了電極材料的循環性能.此外,采用大離子半徑的稀土元素進行摻雜,從某種程度上擴展了正極材料中的三維擴散通道,促進了Li離子的遷移.Sun等[17]研究了LiMn2–xRExO4(0 ≤x≤ 0.01,RE=La,Ce,Nd,Sm)的正極材料.研究結果表明,與純的LiMn2O4相比,稀土摻雜后的材料循環性能和倍率性能均有顯著提高.不僅是LiMn2O4正極材料,Zhang等[18]還在LiFePO4中用La替代Li實現了可逆容量、電導率和Li離子遷移率的顯著提高.在三元材料的摻雜研究方面,實驗表明Li[Ni1/3Co1/3Mn1/3]1–xRexO2(0 ≤x≤ 0.04,Re=La,Ce,Pr)具有較高放電能力和較好的循環性能,其原因是稀土元素的摻入成功抑制了循環過程中的電荷轉移[19].
層狀的富鋰錳基固溶體因其具有高容量、高電壓、成本低以及環保等優點,是具有潛力的下一代鋰離子動力電池正極材料.富鋰錳基正極在不斷的充放電過程中存在的循環性能差、電壓衰減、向尖晶石結構的不可逆轉變、不可逆容量大等問題,與富鋰錳基材料中的主要成分Li2MnO3的活化過程密切相關[20].不僅如此,Li2MnO3在深度脫鋰的過程中會發生釋氧.釋氧對于電池的安全來說是一個不容忽視的隱患,它會導致電池的鼓包甚至是爆炸[21].為了提高Li2MnO3的穩定性和電化學性質,人們提出了摻雜改性的方案.Gao等[22]通過第一性原理計算研究發現Mn位Mo摻雜能夠減小帶隙,促進Li離子輸運以及起到抑制釋氧的作用.此外,我們以前的理論研究表明,Li2MnO3利用P摻雜可以抑制氧缺陷,阻止氧氣的形成,并且通過抑制由層狀材料到尖晶石結構的相變,從而提高結構的穩定性[23].
上述關于Li2MnO3的研究表明,摻雜可以有效改善電池正極材料的性能.盡管Li2MnO3的摻雜研究很多,但有關稀土摻雜的研究工作尚未見報道.由于稀土摻雜在鋰離子電池電極材料的改性中能起到良好的效果,并且采用理論模擬的方法能夠方便地預測材料的性能[24],所以在本研究工作中,采用第一性原理計算的方法研究稀土元素(La,Ce,Pr,Sm)摻雜Li2MnO3的原子結構、電子結構以及離子遷移動力學性質,從理論上預測稀土摻雜對Li2MnO3的改性效果.
2 計算細節和方法
采用基于密度泛函理論的第一性原理平面波贗勢方法,所有的數據模擬在Viennaab initiosimulation package軟件包中進行[25].原子實和價電子之間的相互作用通過投影綴加平面波贗勢來描述[26].平面波的截斷能設置為550 eV.電子間相互作用的交換關聯能采用廣義梯度近似(GGA)的Perdew-Wang(PW91)[27]方案處理.由于廣義梯度近似不能準確地描述強關聯體系的電子性質,所以本文采用GGA+U[28]的方法來弛豫離子和晶胞.其中,Mn的有效U值取為5.0 eV[29,30],該U值用于處理Mn的強局域化的d電子.對于稀土元素,可能含有d電子或者f電子,因此在分析原子的電子構型的基礎上,我們對La的d電子和Ce,Pr,Sm的f電子加U值,且La,Ce,Pr和Sm的有效U值分別取7.50,5.30,6.05和7.05 eV[31].布里淵區的數值積分采用Monkhorst-Pack方法[32]劃分積分網格為3 × 3 × 3.因為體系具有磁性,所以所有的計算都考慮自旋極化.對費米能級采用高斯展寬,展開寬度取為0.02 eV.對于體系的結構弛豫,保證作用在每個原子上的Hellman-Feynman力小于0.01 eV/?.此外,采用彈性能帶法[33,34]搜索Li離子的遷移路徑,并計算遷移勢壘.
3 結果與討論
3.1 稀土摻雜的Li2MnO3的原子結構
Li2MnO3是一種空間群為C2/m的富鋰錳基正極材料.其中,Li占據晶體的4h,2c和2b位置,O占據了8j和4i位置,Mn占據了4g位置.2b位置的Li離子在過渡金屬層中,4h和2c位置的Li離子在Li層中[35].我們對2 × 1 × 2超胞結構進行優化,優化后的晶格參數列于表1中,其中,2a=10.030 ?,b=8.670 ?,2c=10.191 ?,與實驗值非常接近[34].隨后,對未摻雜Li2MnO3的完美結構進行稀土摻雜.所有的摻雜都是將一個Mn原子替換成稀土原子(La,Ce,Pr,Sm),如圖1所示.結構優化之后,我們發現晶胞的形狀并沒有改變,依然保持未摻雜時的結構,但晶格常數明顯增大,同時晶胞的體積也增加,如表1所列,顯然這是因為稀土元素的離子半徑比Mn離子大所導致的.此外,對比四種稀土元素摻雜后的晶格體積可以發現,La摻雜的Li2MnO3體積變化最大,而Sm摻雜的體系體積變化最小.盡管實驗上目前還缺少Li2MnO3稀土摻雜的結構數據,但可以發現有關鋰離子電池正極材料LiMn2O4的結果.有趣的是,Sun等[17]以及Iqbal和Ahmad[9]的實驗結果表明LiMn2O4進行稀土摻雜后,晶格常數相比于未摻雜時的減少了.他們給出的解釋是由于稀土離子替位Mn離子后形成的RE—O鍵比Mn—O鍵更強,因此RE—O鍵長比Mn—O鍵長更短,從而導致了晶格常數的減小.然而,根據我們獲得的Li2MnO3的晶格優化結果,我們推斷稀土離子替換Mn離子并不能減小稀土離子與O離子之間的鍵長,所以實驗上觀測到的LiMn2O4由于稀土摻雜造成的晶格常數減小還存在其他的影響因素,此方向值得進一步研究.
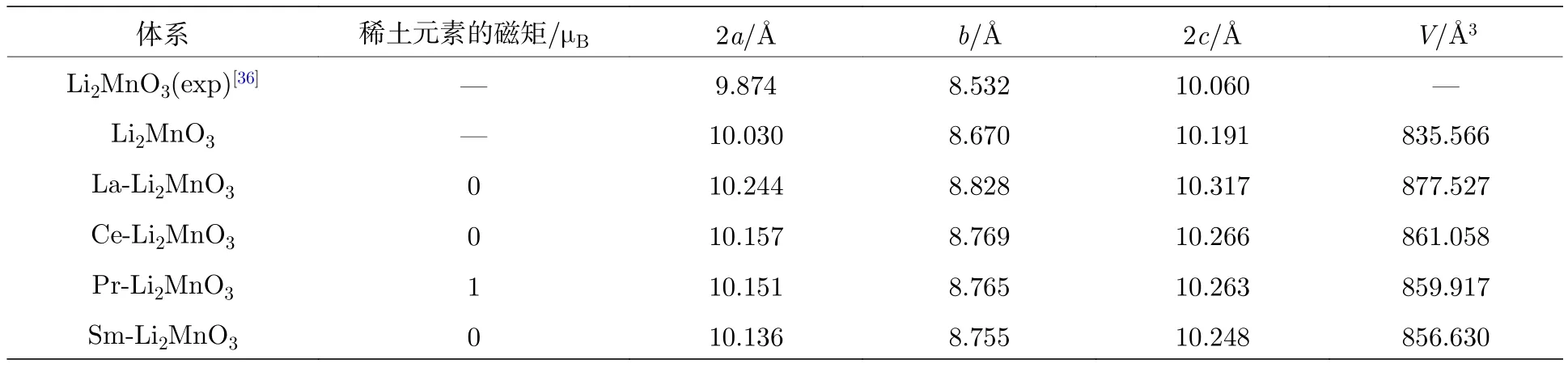
表1 未摻雜與稀土摻雜的Li2MnO3的晶格常數、超胞體積與稀土元素的磁矩Table 1.Lattice constants,volume of supercell,magnetic moment of rare-earth atom of Li2MnO3 without and with rareearth doping.
為了了解稀土摻雜對Li2MnO3結構鍵長的影響,表2列出了稀土摻雜的Li2MnO3優化之后的有關稀土離子與最近鄰O離子之間的詳細鍵長.為了更好地進行比較,表2還給出了未摻雜Li2MnO3中MnO6八面體的鍵長.在未進行稀土摻雜時,MnO6八面體中的六條Mn—O鍵長幾乎相等,其中四條鍵長為1.941 ?,另外兩條分別1.934和1.949 ?.當Mn離子被La離子替換后,La—O之間的鍵長明顯擴大,六條鍵長每兩條相等,三組鍵長分別為2.374,2.385和2.401 ?.受到La摻雜的影響,La近鄰的MnO6八面體中的Mn—O鍵長也發生了變化,鍵長范圍在1.910—1.967 ?之間.當Mn離子被Ce離子替代后,Ce—O鍵長分別為2.198,2.215和2.218 ?,其近鄰的MnO6八面體中Mn—O鍵長范圍在1.931— 1.955 ?之間.當Pr和Sm離子替換Mn離子之后,Pr—O和Sm—O的鍵長分別在2.193—2.211 ?和2.170—2.183 ?范圍內,近鄰MnO6八面體中Mn-—O鍵長變化范圍分別為1.940—1.955 ?和1.916—1.964 ?.計算數據表明,當Li2MnO3中摻入稀土元素后,稀土離子與O離子之間的鍵長與未摻雜結構中的Mn—O鍵長相比都顯著增大,從而擴大了整個晶體的晶格常數.
3.2 稀土摻雜的Li2MnO3的電子結構性質
圖2是未摻雜Li2MnO3的電子態密度圖.從圖2中可以看出,Li2MnO3是半導體材料,計算帶隙約為1.60 eV.此外,計算數據顯示,Mn在體系中呈現+4價,磁矩為3 μB.根據晶體場理論,Mn4+離子中的3個電子占據Mn-3d軌道中的t2g軌道,eg軌道為全空的狀態.因為Mn4+離子很穩定,電子不易失去,所以在脫鋰過程中,Mn的價態保持不變,O會補償電荷,從而使結構不穩定.圖3展示了La,Ce,Pr和Sm摻雜后的Li2MnO3的總態密度與稀土離子的投影態密度.由圖3(a)可以看出,當摻入La時,Li2MnO3由半導體轉變為金屬性質,有電子態穿過費米能級.原因是由于La原子的價電子構型為5d16s2,最外層有3個電子,當摻入Li2MnO3后形成+3價的離子La3+.而Li2MnO3中的Mn離子是+4價的離子.因此在這種非等價離子替換的情況下,部分O的電子態由原來的占據態轉變為非占據態,從而形成金屬性的電子結構,如圖3(a)中的O原子的投影態密度所示.與之不同的是,Ce和Pr摻雜后的Li2MnO3的電子態密度圖與未摻雜情況下的非常相似,都顯示出半導體的特性,如圖3(b)和圖3(c)所示.從相似的電子態密度圖上可以判斷出,Ce和Pr摻雜之后都形成了+4價的離子態,與Mn4+的價態相等,是等價態離子替換,從而沒有改變本質的導電性.為了進一步證實,我們分析了摻雜后稀土離子的磁矩,列于表1中.Ce最外層電子排布為4f15d16s2,當形成+4價離子時,外層電子全部失去,導致磁矩為0.類似地,Pr原子外層電子排布是4f36s2,形成+4價離子后剩余一個f電子,總磁矩為1 μB.因此,我們對價態的分析與磁矩計算結果是相符的.盡管Ce和Pr摻雜的Li2MnO3表現為半導體特性,但帶隙較未摻雜的Li2MnO3變小了,帶隙分別降為1.30和1.40 eV.
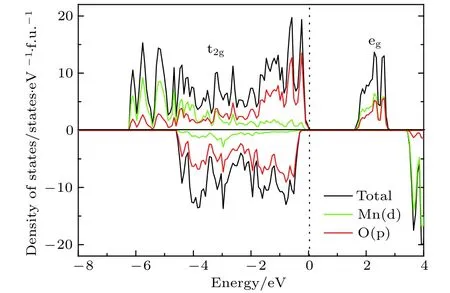
圖2 未摻雜Li2MnO3的電子態密度圖Fig.2.Density of states of Li2MnO3 without doping.
圖3(d)顯示的是Sm摻雜的Li2MnO3的電子態密度.顯然,該電子態密度依然保持了半導體的特性,但帶隙減少為0.91 eV.從總的態密度圖上可以看出,帶隙減小的主要原因是在費米能級附近出現了一個較為局域的帶隙態.為了進一步分析該帶隙態,我們給出了Sm的f軌道的分波態密度,同樣顯示于圖3(d)中.分波態密度結果表明帶隙態幾乎是由Sm的f軌道貢獻的.雖然影響帶隙減小的帶隙態相對比較局域,但在某種程度上對于體系的導電性起到增強的作用.
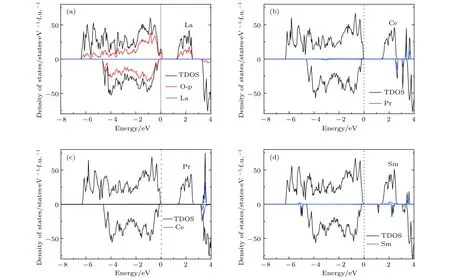
圖3 稀土元素(a)La,(b)Ce,(c)Pr,(d)Sm摻雜的Li2MnO3的電子態密度Fig.3.Density of states of Li2MnO3 with(a)La,(b)Ce,(c)Pr,and(d)Sm doping.
3.3 稀土摻雜的Li2MnO3中的Li離子遷移動力學
由于Li離子在電極化合物中的遷移率是體現可充電鋰離子電池倍率性能的一個關鍵因素,因此研究Li離子遷移的能壘對鋰離子電池的應用至關重要.在稀土摻雜的Li2MnO3中,因為Ce,Pr和Sm都是相同的價態,所以它們對于Li離子遷移的影響是類似的.對比表1中Ce,Pr和Sm摻雜后的Li2MnO3的晶格常數以及表2中Ce—O,Pr—O和Sm—O鍵長可以發現,Ce,Pr和Sm摻雜產生的結構影響相差很小.因此,我們選取具有代表性的La和Ce元素摻雜的Li2MnO3體系,研究其中的Li離子遷移動力學.
參考Xiao等[37]報道的關于Li2MnO3中Li離子遷移路徑的分析,并且結合稀土元素的摻雜位置,本文中我們定義了9個Li離子的位置,示于圖1中.因為超胞的產生是原胞在a方向擴了一倍,根據周期性邊界條件,以6號,4號和3號Li原子為鏡面,整個晶胞左右對稱.因為La/Ce的摻入,體系不再對稱,即使相同的wyckoff位置也具有不同的遷移類型.為了便于描述,相對于摻雜的稀土離子而言,我們把靠近稀土離子一邊的Li位定義為近鄰位,包括1,2和5號位; 把遠離稀土離子一邊的Li位定義為遠端位,包括1',2'和5'號位,剩下的6,3和4號位則作為中間位.其中,6是2b位,3和4都是2c位,1,2,5(或1',2',5')都是4h位.為了全面分析遷移路徑,我們總共考慮了11條遷移路徑的能壘,如表3所列,并與未摻雜時的遷移路徑進行比較.
Li離子在La摻雜的Li2MnO3中的遷移情況分成兩類,如圖4所示,左邊一列圖4(a),4(b),4(d),4(f),4(h),4(j)為近鄰位的Li離子的遷移勢壘,右邊一列圖4(c),4(e),4(g),4(i),4(k)是遠端位的Li離子的遷移勢壘.圖4中遠端位的離子遷移勢壘范圍為0.36—0.53 eV,與未摻雜時相同wyckoff位的結果(0.51—0.84 eV)[37]相比,有了明顯降低.這主要是因為Li離子遷移勢壘的大小與過渡金屬層之間的空間密切相關.相比于未摻雜的Li2MnO3,La摻雜后c方向的晶格常數明顯增加,從而拉大了遷移空間,減小了遷移勢壘,使得Li離子的遷移更容易發生.與遠端位的離子遷移情況不同,近鄰位的Li離子遷移的勢壘變化范圍非常大,具體為0.01—1.03 eV.我們推斷如此大的勢壘變化與La摻雜之后的局域結構變化大有關.為了證實這一點,我們分析了La離子周圍的局域結構.結果表明,La摻入后形成了La—O鍵,其鍵長比Mn—O鍵長長,會局域性地壓縮Li層上下O與O之間的層間距.我們的數據顯示,遠端位的Li層上下O與O之間層間距大約為2.817 ?,而稀土離子正下方的O與O層間距只有2.724 ?.因此圍繞著La周圍的離子遷移,勢壘很大,如6-1路徑的0.87 eV和1-5路徑的1.03 eV.從上面的結果可以得出,一方面,稀土摻雜整體上可以降低Li離子的遷移勢壘,提高遷移速率; 另一方面,稀土離子的摻入改變了局域結構,使得周圍的勢能面起伏更大,不利于Li離子的遷移.所以,稀土離子的摻雜濃度對于Li離子的遷移有著重要的影響,需要進一步優化.值得注意的是,我們計算中只將1個Mn原子替換成稀土離子,計算得到的摻雜比例為6.25%.實驗上,Sun等[17]對于LiMn2O4的摻雜比例小于1%,三元材料Li[Ni1/3Co1/3Mn1/3]1–xRexO2(0 ≤x≤ 0.04)的稀土摻雜比例則小于4%[19].盡管我們模型中的稀土摻雜比例略大于實驗的結果,但在同一量級上.因此,我們理論計算的結果與實驗具有可比性.此外,對于1-2,1-3,1-4三條遷移路徑,其對應的能量勢壘呈現出明顯的方向性,即由最靠近稀土離子的Li位(1號位)向其他的Li位遷移,勢壘較低,反之,從其他Li位向1號Li位遷移,勢壘極大.這說明Li離子傾向于向遠離稀土元素的方向遷移.進一步地,在考慮實際遷移過程中,Li離子將會繞過稀土摻雜位而進行遷移.
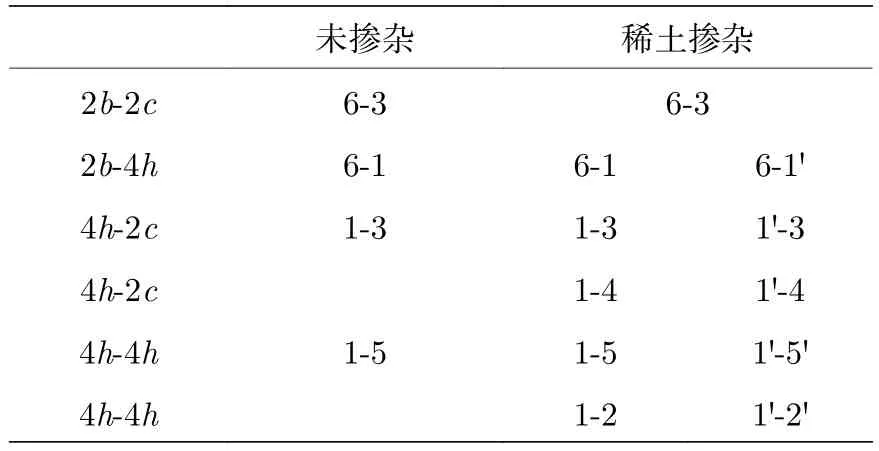
表3 兩種近鄰結構中的Li離子遷移路徑Table 3.Diffusion paths of Li ions in two different structures with neighboring Li ions.

圖4 Li離子在La摻雜Li2MnO3中的遷移勢壘,所有的遷移路徑與表3中所列的一致Fig.4.Diffusion energy barriers of Li ions in La-doped Li2MnO3.All diffusion paths are consistent with those listed in Table 3.
Li離子在Ce摻雜的Li2MnO3中遷移與La摻雜的情況類似,遷移勢壘如圖5所示.同樣的wyckoff位的遷移,遠端位的遷移勢壘變化范圍為0.31—0.69 eV,低于未摻雜的情況(0.51—0.84 eV)[37].在遠離稀土離子處遷移勢壘呈現出不同程度的減小,原因是O和O層間距離較未摻雜時增加,減小了Li離子遷移時的阻礙.對于近鄰位的離子遷移,勢壘變化范圍為0.31—0.89 eV.其中1-5路徑的遷移勢壘較未摻雜時(0.74 eV)有所增加,但較La的情況要小.因此在Li2MnO3中Li離子的遷移性質方面,Ce摻雜的影響比La摻雜的要小.具體原因可以從兩個方面考慮: 一方面,依據前面討論的結構信息可以發現La摻雜對晶格的影響要比Ce摻雜的大,因此La的摻雜導致的勢能面畸變相應也會比Ce的大,這將會影響Li離子的遷移勢壘; 另一方面,Ce摻雜Mn位是等電子摻雜,摻雜位上的電荷變化不大,而La摻雜Mn位屬于少電子元素摻雜(La是+3價,Mn是+4價),摻雜位可以近似認為是一個電子受主,因而對Li離子的束縛更大,導致Li離子的遷移更加困難,這與Li摻雜的ZnO中的H的遷移具有一定的相似性[38].
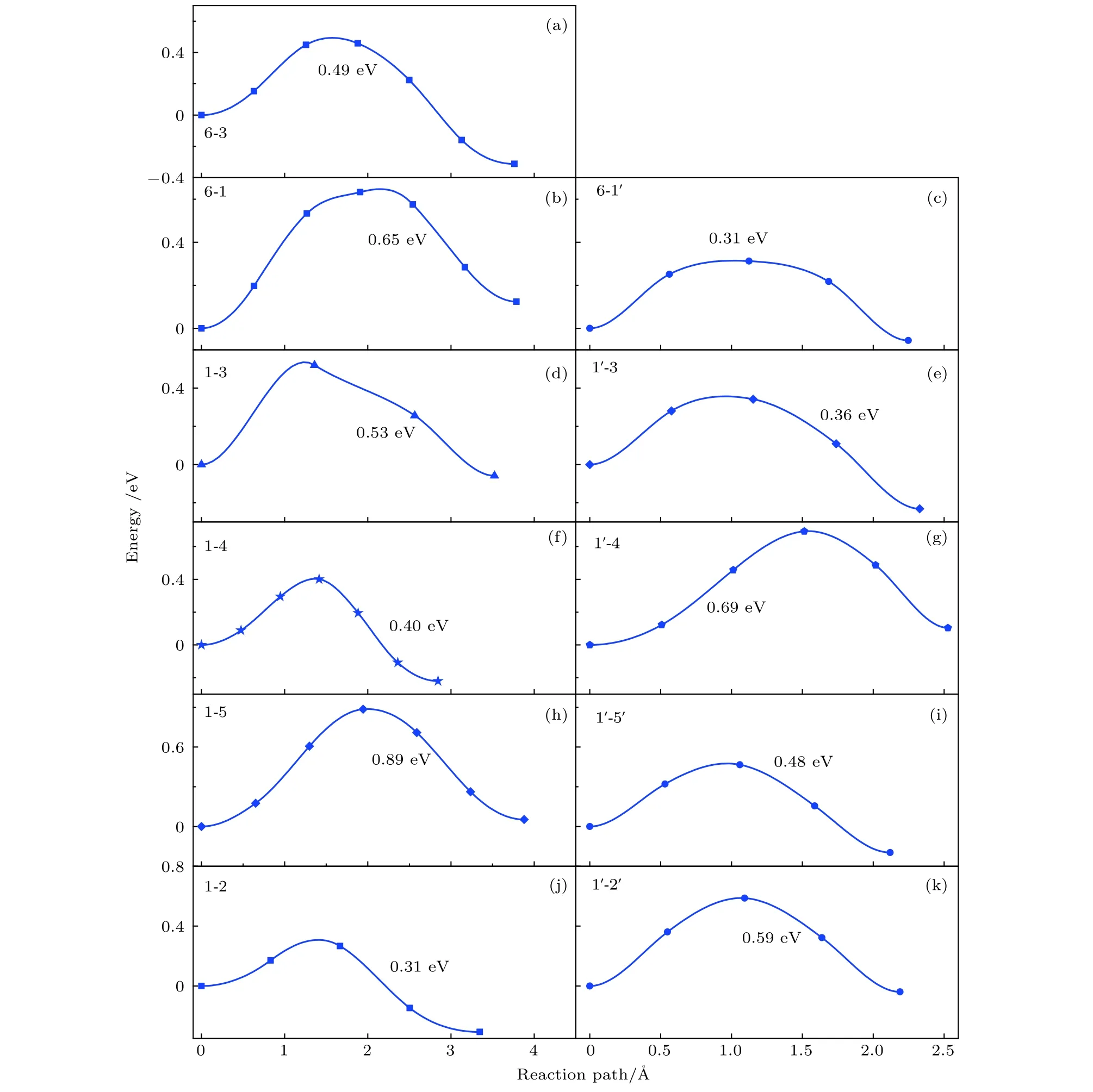
圖5 Li離子在Ce摻雜Li2MnO3中的遷移勢壘,所有的遷移路徑與表3中所列的一致Fig.5.Diffusion energy barriers of Li ions in Ce-doped Li2MnO3.All diffusion paths are consistent with those listed in Table 3.
4 結 論
本文采用第一性原理的方法研究了稀土(La,Ce,Pr,Sm)摻雜的鋰離子電池正極材料Li2MnO3的結構、電子結構和離子遷移性質.結果表明,我們所考慮的稀土元素摻雜均增加了Li2MnO3的晶格常數和晶胞體積,其中La摻雜的晶格常數增加最大,而Sm摻雜的增加最小.La摻雜使得Li2MnO3從半導體轉化為金屬性質,Ce,Pr,Sm摻雜則使得Li2MnO3仍然保持半導體性質,其中Ce,Pr摻雜略微減小了帶隙,而Sm則使帶隙從未摻雜時的1.60 eV減小為0.91 eV.從La和Ce摻雜的Li2MnO3中Li離子遷移的情況看,在遠離稀土離子處遷移勢壘呈現出不同程度的減小,原因是O和O層間距離較未摻雜時增加,減小了Li離子遷移時的阻礙; 而在靠近稀土離子處遷移勢壘起伏較大,特別是在稀土離子最近鄰處的Li離子遷移勢壘明顯增大,其原因主要是摻雜的稀土離子附近的局域結構變化很大,從而導致勢能面起伏很大.與Ce摻雜比較,La摻雜造成的離子遷移勢壘的變化程度更大.此外,本文的結果還表明稀土離子附近的Li離子遷移呈現出方向性,使得Li離子傾向于向遠離稀土離子的方向遷移.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50