線粒體ND4基因11777C>A突變致Leigh綜合征1例家系分析
2019-08-19 11:07:40岳璇劉曉鳴陳嬌桑艷劉莉
山東醫藥 2019年20期
關鍵詞:基因突變
岳璇,劉曉鳴,陳嬌,桑艷,劉莉
(徐州市兒童醫院,江蘇徐州221000)
Leigh綜合征是由于線粒體DNA突變使細胞色素C氧化酶(COX)及丙酮酸脫氫酶復合物缺乏導致細胞氧化磷酸化功能障礙、ATP不能合成而產生不同臨床癥狀的神經疾病,又稱亞急性壞死性腦脊髓病[1]。Leigh綜合征多于嬰兒早期發病,對中樞神經系統具有極大破壞作用[2]。臨床表現多以進行性精神運動損害、喂養困難、呼吸異常為主,其影像學特征是基底節、丘腦、腦干和脊髓的對稱性病變。Leigh綜合征呈線粒體遺傳(又稱母系遺傳,由線粒體基因突變引起)、常染色體隱性遺傳、常染色體顯性遺傳、X-連鎖遺傳(后三種為核基因突變引起),具有臨床表型和遺傳異質性,故易造成漏診或誤診。國內外研究表明,線粒體呼吸鏈5種酶復合物(Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ)缺陷、丙酮酸脫氫酶復合物缺陷及線粒體tRNA突變等均可引起Leigh綜合征[3]。國外研究顯示,核基因突變導致的Leigh綜合征占90%,而線粒體DNA突變導致的Leigh綜合征只占10%。Leigh綜合征發病相關基因多為ND亞基因和COX基因[4]。國外研究發現并證實ND亞基因中ND2、ND3、ND5、ND6突變與Leigh發病具有直接關系[5],但國內關于ND基因突變的報道較少。現將1例線粒體ND4基因11777C>A突變導致Leigh綜合征患兒及其家系資料報告如下。
1 資料分析
患兒7歲余,男。患兒為第一胎、第一產,智力正常,自幼運動發育智力較同齡兒童稍落后,1歲10個月能獨走,2歲會叫“爸爸、媽媽”,3歲左右會說簡單句子。3歲半患兒出現眼斜視,平時喜靜不喜動。3個月前家長發現患兒說話含糊不清,聲音小,未予重視。半個月前出現吃固體食物困難,進而出現吃流質食物困難,伴嗆咳,擬診“支氣管肺炎”輸液治療10 d,癥狀逐漸加重,出現流涎,為排除食管異物,擬行食管鏡入院。入院后查體:神志清,呼吸淺快,偶有嘆氣樣呼吸,面部表情僵硬,轉頭及聳肩無力,左眼外斜視,雙眼眼球震顫陽性,流涎明顯,說話聲音小,吐詞不清,咽反射消失,伸舌居中,鼻唇溝對稱,心肺查體無特殊,四肢肌張力稍高,肌力四級,膝反射活躍,巴賓斯基征陰性。
入院后查血氨、血乳酸正常,血常規、肝腎功能、電解質、心肌酶、免疫球蛋白正常,血尿遺傳代謝篩查均陰性。心臟彩超未見異常,腦電圖示正常腦電圖。腦脊液細胞數及生化正常,腦脊液寡克隆蛋白陰性,自身免疫性腦炎抗體陰性,血沉、自身免疫指標均正常。肌電圖示四肢近端肌肉運動單位時限縮短,四肢神經運動和感覺傳導功能正常。頭顱MRI示右側尾狀核體部、小腦半球及腦干可見多發片狀稍長T1、稍長T2、稍高flair信號,DW1呈不均勻高信號,以腦干及左側小腦半球明顯,腦干異常信號呈輕度強化(圖1)。脊髓MRI未見明顯異常。患兒弟弟行頭顱MRI檢查示小腦半球、腦干、尾狀核丘腦等異常信號(圖2)。根據患兒臨床表現,結合頭顱MRI檢查結果,認為符合Leigh綜合征表現。給予能量、輔酶Q10支持治療。患兒出現咳嗽、流涎、吞咽困難后轉至重癥監護室,后家長放棄治療3 d后死亡。
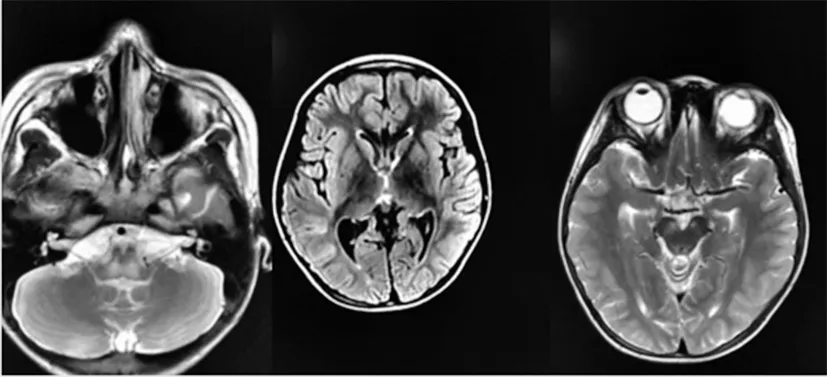
圖1 患兒頭顱MRI影像
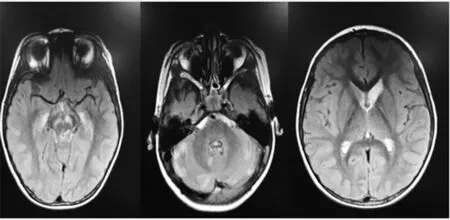
圖2 患兒弟弟頭顱MRI影像
通過DNA芯片結合直接行線粒體測序,在患兒線粒體基因發現了18個突變位點,其中17個突變位點為已報道的多態性位點,只有DN4基因11777C>A是已證實的致病性突變,并有相關報道。患兒ND4基因11777C>A,突變率>62.36%;患兒弟弟ND4基因11777C>A,突變率>67.73%;患兒母親ND4基因11777C>A,突變率>6.40%。患兒弟弟已發病,口服輔酶Q10等治療。患兒母親尚無臨床表現。患兒家系圖見圖3。

圖3 Leigh綜合征患兒家系圖
2 討論
Leigh綜合征是一種多發于嬰幼兒期的線粒體腦病之一,成人罕見。該病臨床表現復雜多樣,其中對神經系統損害最為嚴重,主要表現為精神運動發育落后、眼球震顫、眼肌麻痹、共濟失調、肌張力障礙、肌無力等。因Leigh綜合征臨床表現復雜,涉及多系統損害,臨床醫生對該病認識不足,使得該病的診斷存在一定困難。目前Leigh綜合征尚無公認的診斷標準,主要依據臨床表現、神經影像學特征等。本例患兒為男性,7歲余,病情進展緩慢,出生時無異常,自幼運動發育落后,逐漸出現眼斜視、眼球震顫、吞咽困難、言語不清、肌張力障礙、肌無力,同時伴有面部僵直、毛發增多等,涉及多系統損害,其中對神經系統損害最大,符合線粒體疾病的臨床表現。該患兒已因呼吸衰竭死亡。患兒弟弟現4歲余,臨床表現和病程經過與先證者類似,現已出現眼斜視;與先證者相同,頭顱MRI表現為基底節、腦干、小腦等部位長T1、長T2信號。大多數Leigh綜合征患者血或腦脊液中乳酸水平升高,但這不是特異性指標,有研究發現25%的患者血或腦脊液中乳酸水平正常[6~8]。本例患兒查血乳酸正常,未做腦脊液中乳酸檢查。該患兒未做肌肉活檢,行肌電圖檢查示存在肌源性損害同時伴有面部神經源性損害,證實了Leigh綜合征損害累及多個系統。患兒行血尿遺傳代謝疾病檢測,排除了氨基酸、有機酸、脂肪酸代謝病及炎癥、中毒和維生素B1缺乏、血管性疾病[9,10]。
首例線粒體基因突變疾病于1962年被首次報道[11],學者推測其可能致病機制為線粒體呼吸鏈電子傳遞缺陷,導致細胞ATP合成受阻。這一學說隨后也得到了Holt、Wallace等[12]的證實并進一步完善。Bourgeront等[13]發現并首次報道核基因缺陷同樣也可導致線粒體呼吸鏈功能障礙并引起不同的臨床表型。因此,導致線粒體疾病的基因突變不僅包括核基因突變和核修飾基因突變,還包含線粒體自身的tRNA、rRNA和蛋白編碼基因的突變。由于線粒體氧化磷酸化受自身基因和核基因的雙重調控,故線粒體DNA有其獨特的遺傳模式。第一類是核基因遺傳模式,可分為常染色體顯性遺傳、常染色體隱性遺傳、X-連鎖遺傳。第二類屬于母系遺傳。母系遺傳是線粒體DNA的主要遺傳方式,線粒體DNA在復制和分離過程產生突變,當子代突變的線粒體DNA達到一定比例或閾值后就會發病。研究發現,由于突變的異質性、個體差異及不同器官對線粒體突變的敏感性和閾值不同,線粒體疾病可出現多樣的臨床癥狀[14]。
目前認為Leigh綜合征主要由線粒體呼吸鏈酶缺乏及能量代謝障礙所致[15,16]。由于Leigh綜合征與多種核基因和線粒體基因突變相關,其病因診斷困難。研究表明,接近90%的Leigh綜合征患者存在核基因突變,SURF1基因突變常見,其可導致COX缺陷,從而造成呼吸鏈障礙;線粒體DNA突變約占Leigh綜合征的10%,其中以點突變最多見。張堯等[17]對國內28省145例Leigh綜合征患者篩查發現線粒體基因突變陽性率達到8.3%,其中T10191C最多見,突變率與國外報道相近。國外學者[18]曾報道線粒體ND4基因11777C>A突變導致的Leigh綜合征。本研究中1例患兒的家系基因檢測結果顯示,該患兒為母系遺傳,線粒體DNA為其主要遺傳方式,其母親存在ND4基因11777C>A突變,突變率>6.40%,傳下一代(患兒及其弟弟)均攜帶相同致病基因(ND411777C>A),突變率分別為62.36%、67.73%,均來自其母親線粒體基因突變。患兒及其弟弟已先后出現臨床癥狀,現患兒已死亡,但患兒母親目前未有任何臨床表現,考慮與其突變率低有關,或由于突變的異質性、個體差異及不同器官對線粒體突變的敏感性和閾值不同而產生差異。
Leigh綜合征的預后與發病年齡有很大關系,常見的死亡原因是中樞性呼吸衰竭。磁共振灌注成像可以全面評價Leigh綜合征中樞神經系統受累情況,對早期診斷、病情監測及預后評價具有重要意義[19]。DWI、MRS、FAIR及視路DTI可用于檢出Leigh綜合征患者腦部、脊髓及視路病變,并進行量化分析。目前Leigh綜合征尚無有效治療方法,主要是改善臨床癥狀、提高生活質量,治療原則主要是補充線粒體氧化呼吸鏈中的相關輔酶,如各種維生素、輔酶Q10、左旋肉堿等療法。最近,有學者[20]設計了一段針對線粒體DNA突變tRNAlys m.8344A>G和ND5m.13513G>A的motiTALENs基因片段,通過實驗發現,在后續的基因轉錄中,其可修復目標位置缺陷導致的線粒體DNA突變,使線粒體氧化磷酸化功能恢復,這為Leigh綜合征的基因治療開辟了新的思路。
總之,Leigh綜合征病因復雜多樣,涉及線粒體呼吸鏈多種酶復合物,與核基因組和線粒體基因組的多種基因突變相關。本研究發現了線粒體ND4基因11777C>A突變,明確了患者的病因,進一步充實了人類Leigh綜合征致病基因庫,將有助于今后Leigh綜合征家系的遺傳咨詢。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
