超高效合相色譜-串聯質譜手性分離和測定煙草及土壤中茚蟲威對映體
2019-08-13 10:01:38李中皓劉珊珊范子彥邊照陽唐綱嶺鄧惠敏
煙草科技 2019年7期
關鍵詞:煙草
楊 飛,李中皓,王 穎,劉珊珊,范子彥,邊照陽,唐綱嶺,鄧惠敏
國家煙草質量監督檢驗中心,鄭州高新技術產業開發區楓楊街2 號 450001
目前,大約有30%的登記農藥含有1 個或多個手性中心[1]。然而,由于生產技術和成本的原因,大多數手性農藥仍以外消旋體的形式銷售和使用。手性農藥的對映體具有不同的生物活性、毒性及其他性質。通常,手性農藥的活性只存在于1個或少數幾個異構體中,而對生物活性無效或低效的異構體可視為農藥中的“雜質”[2-3]。例如,2,4-滴丙酸的除草活性幾乎全部集中在R-對映體上,其S-對映體幾乎沒有除草活性[4]。
茚蟲威屬于具有噁二嗪結構的氨基甲酸酯類高效殺蟲劑,適用于防治甘藍、辣椒、煙草等作物上的卷葉蛾類、菜青蟲等。其分子結構中含有1 個手性中心,具有1 對對映體(圖1)[5]。有研究表明,茚蟲威的殺蟲活性主要來源于S-異構體[6]。McCann 等[7]、李曉剛等[8]研究了茚蟲威對映體在土壤中的選擇性降解行為,發現土壤理化性質對茚蟲威對映體的降解有很大的影響。在國際煙草科學研究合作中心(Cooperation Centre for Scientific Research Relative to Tobacco,CORESTA)農用化學品咨詢委員會(Agro-Chemical Advisory Committee,ACAC)制定的106 種農用化學品指導性殘留限量表(Agrochemical Guidance Residue Levels,GRL)中,茚蟲威(R-茚蟲威和S-茚蟲威之和)的殘留限量被定為15 mg/kg[9]。
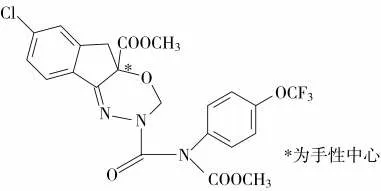
圖1 茚蟲威化學結構Fig.1 Chemical structure of indoxacarb
研究人員主要采用高效液相色譜(High Performance Liquid Chromatography,HPLC),以正相色譜模式對茚蟲威進行手性分離和測定[10-13]。該方法需要消耗大量有毒有害的有機溶劑,而且分析時間較長。近年來,超臨界流體色譜(Supercritical Fluid Chromatography,SFC)引起了研究人員越來越多的關注[14-17]。與HPLC 相比,SFC 以低黏度二氧化碳為主要流動相,且允許使用更高的流速,從而獲得更快的分離速度;同時,SFC 的有機溶劑消耗量低于HPLC。上述優點使SFC 在手性化合物的分析、測定和制備分離中得到了廣泛應用。超高效合相色譜系統(Ultra Performance Convergence Chromatography,UPC2)是一種超高效超臨界流體色譜,所用色譜柱的填料為亞2 μm 顆粒,與使用亞5 μm 顆粒的色譜柱相比,這種色譜柱具有更快的分離速度,而且超高效合相色譜與串聯質譜(Tandem mass spectrometry,MS/MS)聯用后可進一步提高方法靈敏度[18-22]。本研究中基于UPC2-MS/MS 開發了一種環境友好、靈敏快速的手性分析方法,對煙草及其土壤中的茚蟲威對映體進行立體選擇性拆分和測定,旨在為煙草農殘分析提供方法參考。
1 材料與方法
1.1 材料、試劑和儀器
10 個烤煙樣品,全部來自2018 年度國家煙草專賣局農殘普查分析樣品;10 個土壤樣品,分別來自10 個煙葉樣品對應的產地。所有樣品烘干后粉碎,過2 mm(10 目)篩后置于棕色玻璃瓶中,于4 ℃下保存。
外消旋茚蟲威(R-/S-茚蟲威,質量分數均為50%,德國Augsburg 公司);R-茚蟲威(>96.0%,天津阿爾塔科技有限公司);乙腈、乙醇、甲醇、異丙醇和甲酸(色譜純,德國Merck 公司);無水硫酸鎂(MgSO4)、氯化鈉(NaCl)、N-丙基乙二胺(PSA)(AR,上海阿拉丁生化科技股份有限公司);高純氮、高純氬(>99.99%,鄭州源正科技發展有限公司);水為超純水。
ACQUITY 超高效合相色譜儀、ACQUITY TQD 四極桿串聯質譜儀(配電噴霧電離源)、Acquity UPC2Trefoil CEL1 和Acquity UPC2Trefoil CEL2 柱(100 mm×2.1 mm, 1.7 μm)(美國Waters公 司);Chiralcel OD-3 和Chiralcel IG-3 柱(100 mm×3.0 mm, 3.0 μm)(日本Daicel 公司);VX200渦旋振蕩儀(美國Labnet 公司);SG3-30K 高速離心機(德國Sigma 公司);Milli-Q 超純水系統(美國Millipore 公司);AEl63 電子天平(感量:0.000 1 g,瑞士Mettler Toledo 公司)。
1.2 方法
1.2.1 基質匹配標準工作溶液的配制
稱取20 mg 茚蟲威外消旋體標準品于100 mL容量瓶中,用乙腈定容,制得R-茚蟲威和S-茚蟲威質量濃度均為100 μg/mL 的一級混合標準儲備液。移取1.0 mL 一級混合標準儲備液于10 mL 容量瓶中,用乙腈定容,得到R-茚蟲威和S-茚蟲威質量濃度均為10 μg/mL 二級混合標準儲備液。分別移取二級混合標準儲備液1 000、500、200、100和50 μL 至5 個10 mL 容量瓶中,用乙腈定容,制得不同質量濃度的標準工作溶液。分別移取0.2 mL 標準工作溶液和0.2 mL 空白樣品提取液,混合后用乙腈稀釋至1 mL。各基質匹配標準工作溶液中對映體的質量濃度分別為200、100、40、20 和10 ng/mL。所有溶液避光儲存在-20 ℃的冰箱中。
1.2.2 樣品前處理及分析
參考本課題組前期的研究[23]對樣品進行前處理,簡述如下:稱取2 g 樣品置于具蓋離心管中,加入超純水和乙腈,渦旋振蕩;向離心管中加入無水硫酸鎂、氯化鈉、檸檬酸鈉和檸檬酸二氫鈉,繼續渦旋振蕩;移取1 mL 上清液于1.5 mL 離心管中,加入無水硫酸鎂及PSA,渦旋振蕩;取上清液,用0.22 μm 有機相濾膜過濾;取200 μL 濾液,加入800 μL乙腈,混勻后進行UPC2-MS/MS檢測。UPC2-MS/MS 分析條件為:
色譜柱:Chiralcel IG-3 柱(100 mm×3 mm,3.0 μm);動態背壓:13.79 MPa;柱溫:35 ℃;樣品室溫度:10 ℃;補償溶劑:0.1%甲酸的甲醇溶液,流速0.2 mL/min;進樣量:2 μL;流動相流速:2.0 mL/min;流動相:A 相為CO2,B 相為甲醇;等度洗脫:流動相A∶流動相B=92%∶8%;質譜掃描方式:正離子掃描;離子源:電噴霧電離源(ESI);離子源溫度:150 ℃;脫溶劑氣溫度:350 ℃;脫溶劑氣流量:600 L/h;錐空氣流量:60 L/h;停留時間:100 ms;毛細管電壓:4.5 kV;采集模式:多反應監測(MRM);定量離子對(m/z):527.97>149.95(錐孔電壓:38 V,碰撞電壓:20 V);定性離子對(m/z):527.97>202.97(錐孔電壓:38 V,碰撞電壓:38 V)。
1.2.3 加標樣品制備
在優化的條件下,對實際樣品進行分析。各選取1 個煙葉樣品和土壤樣品作為空白樣品。向空白樣品中添加不同量的外消旋體標準儲備液,得到3 個不同濃度的空白加標樣品,其中R-/S-茚蟲威對映體的含量分別為2.5、7.5、15.0 mg/kg。在樣品中加入10 mL 甲醇并渦旋振蕩10 min,混勻后置于通風櫥中在室溫下揮干溶劑。加標樣品按照1.2.2 節方法處理,每個濃度水平重復測定5 次,連續測定3 d,采用基質匹配標準工作曲線進行定量分析。
1.2.4 數據分析
數據采集采用MassLynx V4.1 軟件。平均值、回收率和相對標準偏差(RSD)的計算采用Microsoft Excel 2007 版軟件。
2 結果與討論
2.1 手性分離條件的優化
2.1.1 手性柱的選擇
分別考察了Acquity UPC2Trefoil CEL1、Acquity UPC2Trefoil CEL2、Chiralcel OD-3 和Chiralcel IG-3 等4 種不同的手性柱對茚蟲威兩種對映體的分離情況。Acquity UPC2Trefoil CEL1 和Acquity UPC2Trefoil CEL2 柱均使用改性的多聚糖型固定相,其中Acquity UPC2Trefoil CEL1 柱的硅膠表面涂敷有纖維素-三(3,5-二甲基苯基氨基甲酸酯),Acquity UPC2Trefoil CEL2 柱的硅膠表面涂敷有纖維素-三(3-氯-4-甲基苯基氨基甲酸酯)。Chiralcel OD 柱為多糖衍生物正相涂敷型手性色譜柱,硅膠表面涂敷有纖維素-三(3,5-二甲基苯基氨基甲酸酯)。Chiralcel IG-3 柱為多糖衍生物耐溶劑型手性色譜柱,硅膠表面以共價鍵鍵合有直鏈淀粉-三(3-氯-5-甲基苯基氨基甲酸酯)。實驗結果(圖2)表明:當使用Acquity UPC2Trefoil CEL1 和Chiralcel OD-3 柱時,不能實現茚蟲威對映體的完全分離;使用Acquity UPC2Trefoil CEL2 和Chiralcel IG-3 柱均可實現對映體的分離,但使用Acquity UPC2Trefoil CEL2 柱時,目標物色譜峰信號明顯減弱。綜合考慮目標物分離度及色譜峰形和響應情況,選擇Chiralcel IG-3 柱對茚蟲威對映體進行分離。
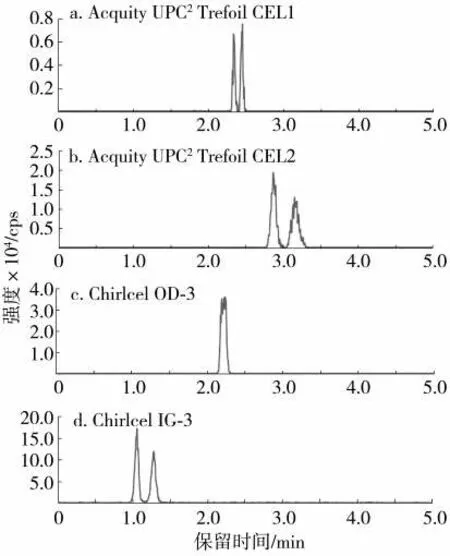
圖2 4 種不同色譜柱上茚蟲威對映體的UPC2-MS/MS 色譜圖Fig.2 Typical UPC2-MS/MS chromatograms of indoxacarb on four different columns
2.1.2 改性劑的確定
超高效合相色譜以超臨界CO2流體為流動相,通常需在超臨界流體中添加少量的有機溶劑(甲醇、乙醇、異丙醇)作為改性劑。改性劑可以改變流動相的洗脫強度,并降低流動相與固定相的硅烷醇基團的相互作用,從而改善色譜峰形[24]。考察了3 種有機改性劑(甲醇、乙醇和異丙醇)對茚蟲威對映體在Chiralcel IG-3 柱上分離效果的影響。結果(圖3)表明,使用甲醇、乙醇和異丙醇均可以實現茚蟲威對映體的基線分離,但當使用乙醇和異丙醇時,茚蟲威對映體的MS 信號明顯減弱且保留時間變長。實驗中還考察了甲酸、乙酸和甲酸銨對茚蟲威對映體拆分的影響,結果表明,加入甲酸、乙酸等并沒有改善對映體之間的分離度和信號響應強度。因此,選擇甲醇作為流動相改性劑。
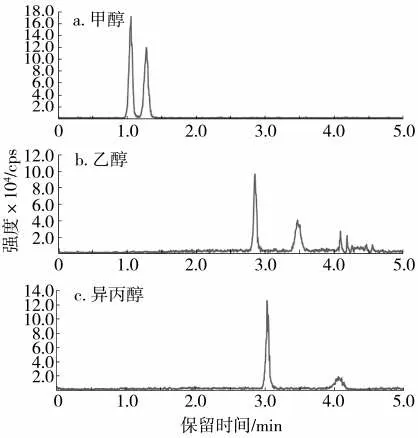
圖3 改性劑對茚蟲威對映體分離效果的影響Fig.3 Effect of modifier solvent on enantioseparation of indoxacarb enantiomers
2.1.3 動態背壓的選擇
動態背壓(Automatic back pressure regulator,ABPR)升高后,流動相密度隨之增加,而目標物的保留時間減小。原因可能是,在高背壓下色譜柱內流動相密度增加,溶劑化能力和洗脫效率均有所提高[25]。分別考察了不同背壓(11.03、12.41、13.79 和15.17 MPa)對茚蟲威對映體分離效果的影響。結果(圖4)顯示,隨著動態背壓的升高,目標物保留時間減少,但是動態背壓對目標物分離效率無顯著影響,茚蟲威對映體的分離度幾乎不變。可見,動態背壓變化在茚蟲威對映體的分離中不起關鍵作用。當動態背壓為13.79 MPa 時,茚蟲威對映體的色譜峰信號響應強度較高。因此,選擇動態背壓為13.79 MPa。

圖4 動態背壓對茚蟲威對映體分離效果的影響Fig.4 Effect of ABPR on enantioseparation of indoxacarb enantiomers
2.1.4 柱溫的確定
對于合相色譜,溫度也會影響目標物的保留時間。隨著柱溫的升高,二氧化碳的密度和黏度降低,對目標物的洗脫能力降低,從而導致目標物保留時間延長[26-27]。在動態背壓13.79 MPa、流速2 mL/min 的條件下,考察了不同柱溫(35~50 ℃)對茚蟲威對映體拆分效果的影響。結果(圖5)表明,隨著柱溫的升高,目標物保留時間增加,但對映體分離度幾乎不變;在柱溫35 ℃時,茚蟲威對映體的色譜峰面積最大,方法靈敏度較高。因此選擇柱溫為35 ℃。
圖5 柱溫對茚蟲威對映體分離效果的影響Fig.5 Effect of column temperature on enantioseparation of indoxacarb enantiomers
2.1.5 茚蟲威對映體的洗脫順序
在優化的條件下,利用R-茚蟲威標準溶液確定茚蟲威兩種異構體在UPC2-MS/MS 分析中的洗脫順序。實驗中,根據先前的研究報道[5]鑒定了茚蟲威對映體的旋光性。結果顯示,茚蟲威對映體在手性柱(Chiralcel IG-3)上的洗脫順序為:S-(+)-茚蟲威(1.05 min)和R-(-)-茚蟲威(1.28 min)。在優化的手性分離條件下,煙草和土壤實際加標樣品的選擇離子色譜圖如圖6 所示。可以看出,在不同的基質中,S-茚蟲威和R-茚蟲威均實現了基線分離,且峰形良好。
2.2 方法評價
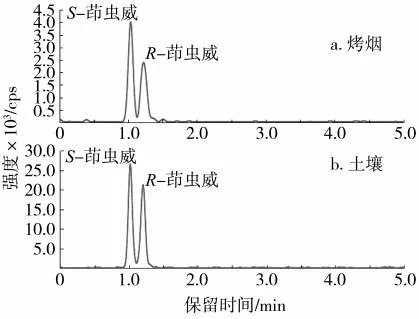
圖6 烤煙和土壤實際加標樣品的色譜圖Fig.6 Chromatogram of actual spiked samples of flue-cured tobacco and soil
2.2.1 基質效應一般認為,液質聯用中的基質效應是由于基質成分和目標物在離子化時相互競爭造成的,表現為離子抑制或離子增強效應,其對儀器的靈敏度和重復性影響顯著[28]。通常采用基質標準曲線斜率和溶劑標準曲線斜率之比(K)來評價基質效應:當K 在0.9~1.1 之間時,基質效應不明顯;當K大于1.1 時為基質增強效應;當K 小于0.9 時為基質減弱效應。結果(表1)表明,在基質不稀釋的情況下,基質效應在0.15~0.39 范圍內,說明在煙草和土壤基質中存在顯著的基質抑制效應。因此,對于煙草和土壤樣品中茚蟲威對映體的測定,均使用基質匹配標準溶液進行定量分析。
基質的稀釋倍數也是影響基質效應的重要因素。實驗中考察了不同稀釋倍數下烤煙和土壤樣品中茚蟲威對映體的基質效應。結果(表1)表明,隨著稀釋倍數的增加,目標物的相對響應強度(各樣品的響應強度與標樣的響應強度之比)也增大,表明稀釋樣品有助于減小基質效應。但是稀釋的作用有限,稀釋倍數過高,則目標物濃度太低,從而影響方法靈敏度。因此,選擇用乙腈將其稀釋5倍后進樣。
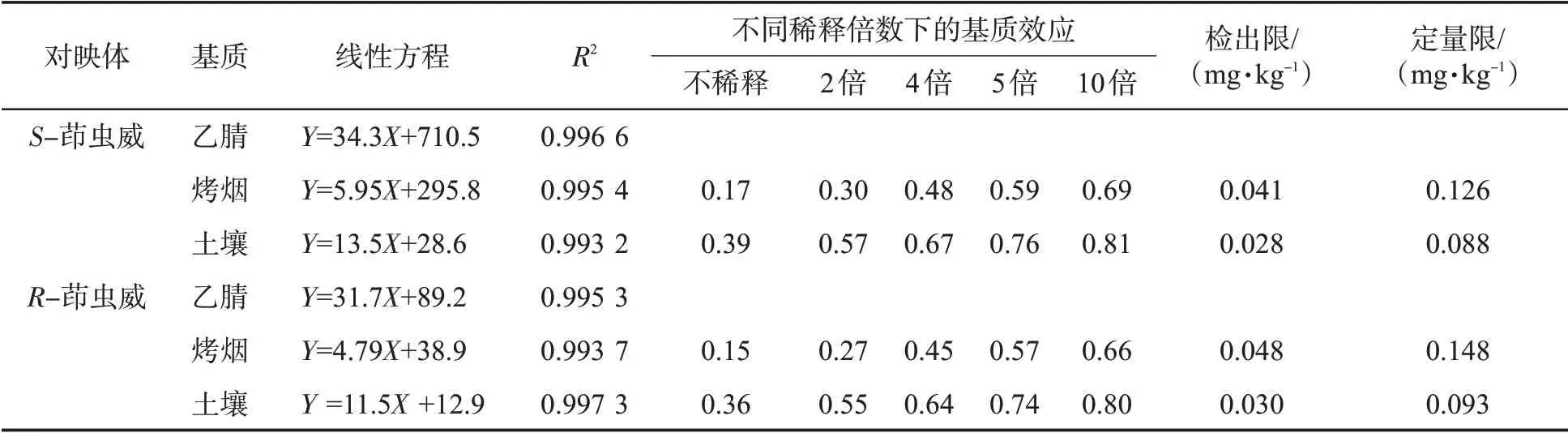
表1 茚蟲威對映體的線性方程、基質效應、檢出限和定量限Tab.1 Regression equation, matrix effect, LODs, and LOQs for enantiomers of indoxacarb
2.2.2 標準曲線、檢出限和定量限
通過繪制茚蟲威對映體色譜峰面積與其濃度的關系圖來生成標準曲線。標準曲線的形式為Y = AX + B,其中A 和B 分別表示為斜率和截距。對于每種對映體,在10~200 ng /mL 的質量濃度范圍內分析標準工作溶液和不同基質匹配的標準工作溶液,以確定該方法曲線的線性。表1 表明,不同基質中各對映體線性關系良好(R2>0.993),可以滿足定量分析的需要。將加標空白煙草和土壤提取物中產生的信噪比(S/N)等于3 和10 時對映體的濃度分別定義為檢出限(LOD)和定量限(LOQ)。由表1 可知,不同基質中LOD 和LOQ 分別為0.028~0.048 和0.088~0.148 mg/kg。
2.2.3 方法回收率及精密度
通過空白樣品加標回收實驗來測試該方法的準確度和精密度。制備了3 個不同濃度的加標樣品,按照1.2.2 節的方法進行前處理,每個濃度的樣品每天重復測定5 次,連續測定3 d。3 個不同濃度的加標樣品中R-茚蟲威和S-茚蟲威的含量為2.5、7.5 和15.0 mg/kg。該方法的精密度由重復性實驗確定,并以RSD 表示。通過比較同一天內加標樣品的回收率的標準偏差來測量日內精密度。通過分析3 個不同日期的加標樣品來確定日間精密度。結果(表2)顯示,在所有濃度水平下均獲得較好的平均回收率(83.7%~96.6%),兩種對映體的日內精密度(RSD)為2.4%~6.1%,日間精密度(RSD)為3.3%~5.8%。
2.3 方法的應用
采用本方法對10 個烤煙樣品和10 個土壤樣品進行了測定,結果顯示:僅有1 個土壤樣品檢出有R-茚蟲威和S-茚蟲威,含量分別為1.05 和0.88 mg/kg,遠遠低于GRL中規定的茚蟲威對映體的殘留限量[9]。該研究結果也與煙草中農藥實際使用情況相吻合。氨基甲酸酯類農藥在煙草中的使用量很小,多年來煙草中該農藥的檢出率一直保持在很低的水平。個別土壤樣品檢出茚蟲威可能是由于輪作所致。另外,由于茚蟲威對映體的降解速率不同,因此,兩種茚蟲威對映體的含量存在差異。
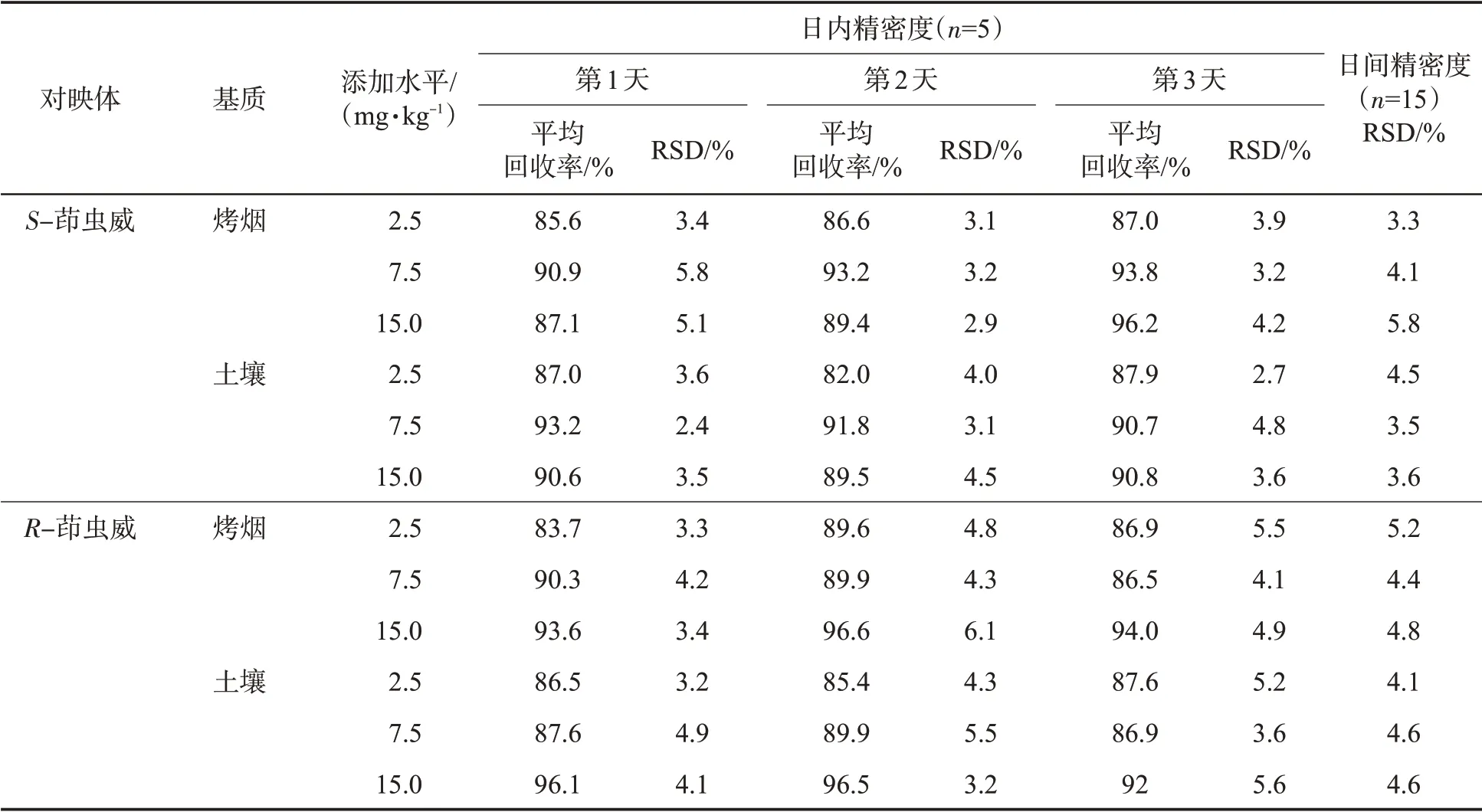
表2 方法的精密度和準確度Tab.2 Accuracy and precision of the proposed method
3 結論
建立了手性分離和測定煙草和土壤樣品中茚蟲威對映體的超高效合相色譜-串聯質譜方法,采用本方法可在5 min 內實現茚蟲威對映體的基線分離。在優化的實驗條件下,茚蟲威對映體的回收率在83.7%~96.6%之間,日內精密度為2.4%~6.1%,日間精密度為3.3%~5.8%。本方法以CO2作為主要流動相,定量結果較為準確可靠。本研究為煙草及土壤中茚蟲威對映體的分離和測定提供了一種綠色、快速、可靠的方法。
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
中國煙草學報(2019年5期)2019-11-14 07:54:12
首都公共衛生(2019年5期)2019-05-21 01:08:34
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
新聞傳播(2016年3期)2016-07-12 12:55:34
當代化工研究(2016年9期)2016-03-20 16:22:08
自動化博覽(2014年6期)2014-02-28 22:32:15
聲屏世界(2014年6期)2014-02-28 15:18:09
西南學林(2013年2期)2013-11-12 12:58:54
中國煙草學報(2012年5期)2012-04-12 06:21:18
