成年灘羊和小尾寒羊皮膚毛囊差異表達miRNA 的篩選
2019-08-12 05:03:32何玉龍莊仁杰周學章吳月紅
中國畜牧雜志 2019年8期
關鍵詞:差異
何玉龍,楊 瑞,莊仁杰,李 勇,周學章,吳月紅*
(1.浙江理工大學生命科學與醫藥學院,浙江杭州 310018;2.寧夏大學西部特色生物資源保護與利用教育部重點實驗室,寧夏銀川 750021;3.浙江理工大學校醫院,浙江杭州 310018)
被毛生長是一個復雜的生理過程,受環境、營養、代謝及基因調控等因素影響,其中基因調控是決定性因素[1]。從基因水平上揭示綿羊被毛生長特點對于改良其品質具有重要價值。MicroRNAs(miRNAs)在調控基因表達方面起重要作用。隨著綿羊相關miRNA 的不斷發現,其在皮膚毛囊中的表達也逐漸受到重視,可能在皮膚毛囊發育中發揮著重要作用[2-5]。
小尾寒羊具有生長速度快、產羔率高、性能遺傳穩定、適應性強等特點,是我國北方地區的優勢綿羊品種[6-7]。利用小尾寒羊生產裘皮的歷史悠久,但隨著市場需求的變化,小尾寒羊逐步向肉用方向選育[8]。在提高小尾寒羊肉產量的同時,加強裘皮性狀品系的選育具有重要意義。但由于目前并未將小尾寒羊毛用性狀作為主要育種目標,導致其毛用性狀未有明顯提高[7,9]。灘羊主要分布在寧夏及其毗鄰地區,是中國唯一生產“二毛裘皮”的綿羊品種[10]。目前,關于灘羊[11]和小尾寒羊[12]皮膚毛囊組織中miRNA 的研究已有報道,但關于兩者皮膚毛囊中miRNA 的比較研究未見報道。為了解灘羊與小尾寒羊在裘皮性能差異的分子機制,本研究通過高通量測序技術比較灘羊與小尾寒羊皮膚毛囊中miRNA 差異,對于從miRNA 水平上解析皮毛性狀形成的分子機理具有重要意義。
1 材料與方法
1.1 樣品采集 灘羊來源于寧夏回族自治區吳忠市紅寺堡區天源良種羊繁育養殖有限公司,小尾寒羊來源于內蒙古察哈爾右翼中旗某養殖戶,分別采集成年灘羊(命名為TY_1)和小尾寒羊(命名為XWHY_1)各3只,每組個體的樣品混合組成樣品池。樣品采集時間為2015 年2—3 月。剪毛消毒后用手術剪刀剪取背部相同位置約3~5 cm2皮膚組織,去除皮下脂肪組織等,剪成約0.5~1.0 cm2小塊,PBS 溶液清洗后投入RNA 保護劑RNAlater(Thermo Fisher Scientific,美國)中。冷藏條件下盡快帶回實驗室,-80℃冰箱保存。
1.2 總RNA 提 取、小RNA 文 庫 構 建 及Solexa 測 序將樣本用液氮充分研磨后,利用Trizol 試劑(Invitrogen,美國)按照產品說明書進行皮膚組織總RNA 的提取,得到的總RNA 分別采用Nanodrop2000(美國)、Qubit 2.0(美國)和Agilent 2100 分析儀(美國)檢測其純度、濃度和完整性。樣品檢測合格后,以總RNA為起始樣品,使用small RNA Sample Pre Kit 試劑盒(Illumina ,美國)構建文庫,首先在small RNA 5'端和3' 端連接接頭,反轉錄合成cDNA,PCR 擴增后利用PAGE 膠電泳篩選目的片段,切膠回收得到的片段即為small RNA 文庫。質控合格后,利用HiSeq2500 進行高通量測序,測序讀長為single-end(SE)50 nt。
1.3 測序數據分析 按照標準的miRNA 測序分析流程進行測序數據統計、小RNA 分類注釋、已知miRNA的鑒定、新發現miRNA 的預測、miRNA 表達量及差異表達分析、差異表達miRNA 靶基因預測及注釋等。
1.4 實時熒光定量PCR 驗證miRNA 測序結果 為驗證測序結果的可靠性,對鑒定出的miRNA 進行實時熒光定量PCR 檢測驗證。以總RNA 為初始模板,隨機選取4 個miRNAs,miRNAs 反轉錄引物為通用頸環序列(GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCA CTGGATACGAC)和目標miRNA 3' 末端6 個堿基的反向互補序列構成。使用Revert Aid First Strand cDNA Synthesis Kit 試劑盒(Thermo Fisher Scientific,美國)按照產品說明書進行cDNA 第一鏈合成,使用SYBR?Premix Ex Taq 試劑盒(TaKaRa,大連)進行qRT-PCR驗證,以U6 為內參基因。miRNA 相對表達量采用2-△△Ct法進行計算。miRNA 熒光定量PCR 引物見表1。所有引物由生工生物工程(上海)有限公司合成。

表1 實時熒光定量PCR 引物
1.5 差異表達miRNA 的篩選、靶基因預測及參與信號通路分析 利用IDEG6 對上述鑒定出的miRNAs 進行表達差異分析,選擇差異表達的miRNAs,分別使用miRanda 和RNAhybrid 軟件進行靶基因預測。對差異表達miRNA 的靶基因進行基因本體數據庫(Gene ontology,GO)富集分析和京都基因與基因組百科全書(Kyoto encyclopedia of genes and genomes,KEGG)通路富集分析。
2 結果與分析
2.1 小RNA 文庫序列統計 如表2 所示,灘羊(TY_1)和小尾寒羊(XWHY_1)樣品分別通過HiSeq 2500 高通量測序,產出的原始數據(Raw reads)數量大于16.35 million(M),對其進行去除低質量數據、5'接頭、3'接頭及污染等處理后,樣品中過濾后序列所占的比例分別為85.68% 和87.38%,質量值≥30 的堿基所占比例(Q30)分別為95.37%和95.40%。
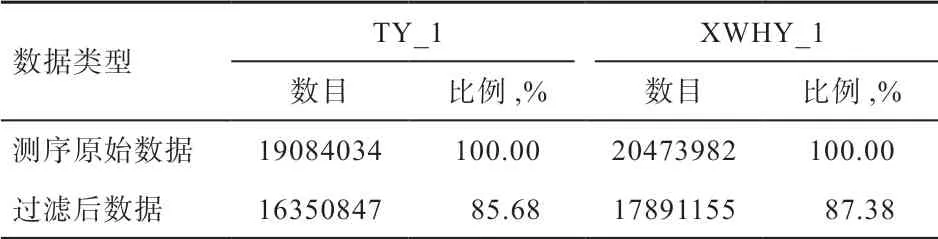
表2 測序數據統計
2.2 sRNA 分類注釋 利用Bowtie 軟件將上述過濾后數據分別與Silva、GtRNAdb、Rfam 和Repbase 數據庫進行序列比對,過濾核糖體RNA(rRNA)、轉運RNA(tRNA)、核內小RNA(snRNA)、核仁小RNA(snoRNA)等非編碼RNA(non-coding RNA,ncRNA)以及重復序列,在TY_1 和XWHY_1 樣品分別獲得包含miRNA 的未注釋的reads(Unannotated reads)10 852 746 和14 135 887,用于后續分析,結果見表3。
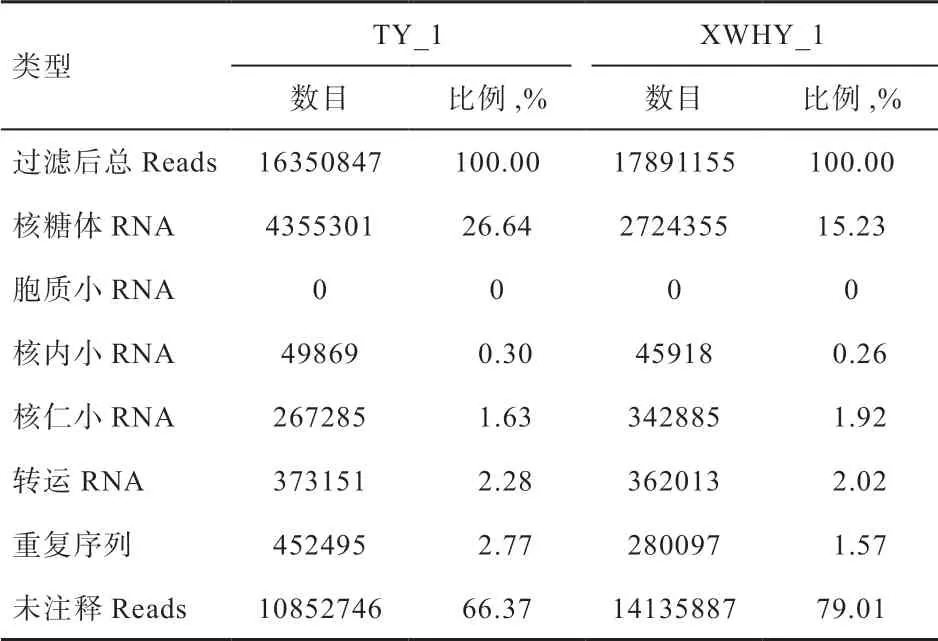
表3 sRNA 分類注釋統計
2.3 與參考基因組比對 利用miRDeep2 軟件將上述未注釋Reads 與參考綿羊基因組(Oar_v3.1)進行序列比對分析,獲取其在參考基因組上的位置信息。如表4 所示,TY_1 和XWHY_1 樣品比對到綿羊基因組的比例分別為52.14% 和51.22%,說明50% 以上的Reads 能比對到綿羊基因組上。
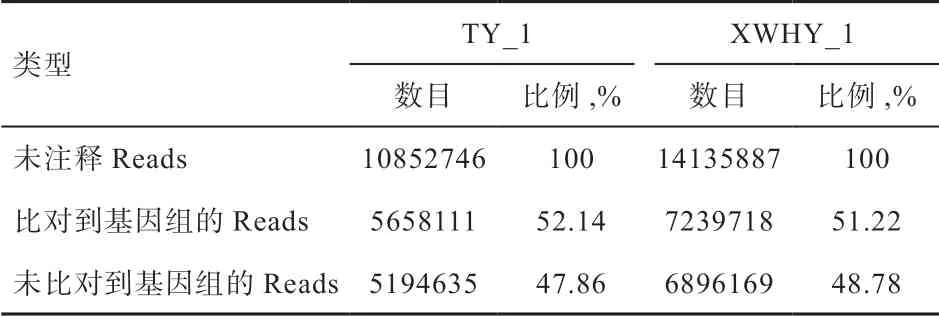
表4 與參考基因組比對信息統計表
2.4 miRNA 的鑒定 對于上述比對到參考基因組的序列,利用miRDeep2 軟件進行已知及新發現miRNA的鑒定。即將18~30 nt 的核苷酸序列比對到miRBase(V21.0)數據庫中特定物種上,鑒定其已知的miRNA。對于未比對到參考基因組的序列,通過堿基數目延伸,進行miRNA 結構預測,獲得新發現miRNA(novel miRNAs)。經過比對共檢測到561 個miRNAs,其中包括138 個已知miRNAs 和423 個新發現miRNAs。
2.5 miRNA 的長度分析 對2 個樣本中鑒定到的138個已知miRNA 和423 個新發現miRNA 進行核苷酸長度統計。如圖1 所示,大部分miRNA 序列長度在20~24 nt,已知miRNA 和新發現miRNA 中所占比例分別為96.38%和95.98%。其中長度為22 nt 的序列所占比例最高,分別為42.75%和51.54%;其次為21 nt 和23 nt,其長度分布符合Dicer 酶切割的典型特征,說明測序結果質量可靠,可用于后續分析。
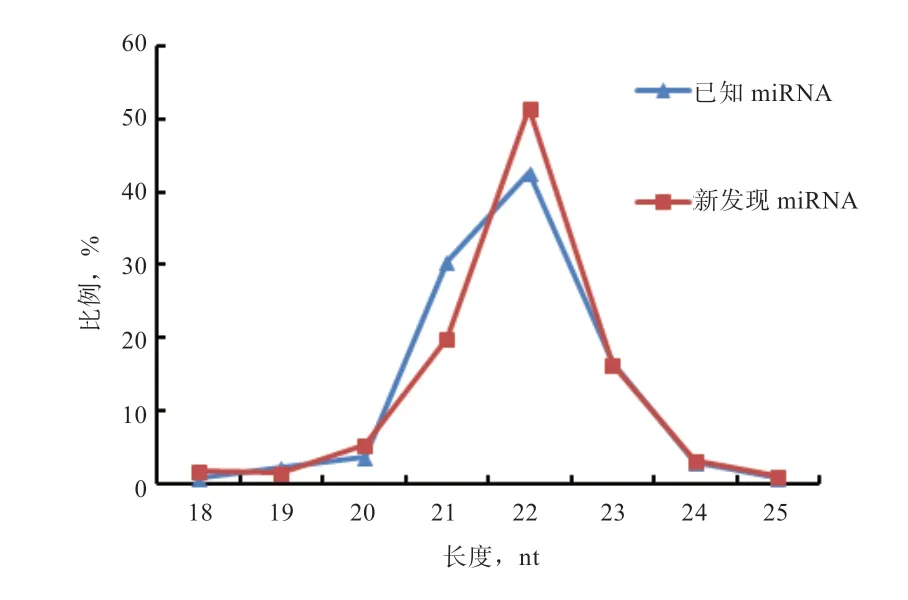
圖1 miRNA 長度分布
2.6 miRNA 表達量分析對TY_1和XWHY_1樣本中鑒定出的miRNA 進行表達量分析,并用TPM 算法對表達量進行歸一化處理。TPM 歸一化處理公式:TPM=Readcount×1000000/Mapped Reads。其中,Readcount表示比對到某一miRNA 的reads 數目;Mapped Reads表示比對到參考基因組上的reads 數目。部分表達量較高的已知miRNA 如表5 所示。oar-let-7 家族在TY_1和XWHY_1 樣本中表達量均較高。

表5 部分高表達已知miRNA 表達量數據(前20 位)
2.7 miRNA qRT-PCR 驗證 隨機選取4 個miRNA,通過qRT-PCR 的方法驗證測序結果的準確性,結果如圖2 所示。通過比較qRT-PCR 與高通量測序的結果發現(表6),這4 個miRNA 在TY_1 和XWHY_1 中表達的變化趨勢基本一致,證明高通量測序結果的可靠性。

圖2 miRNA 的qRT-PCR 驗證分析
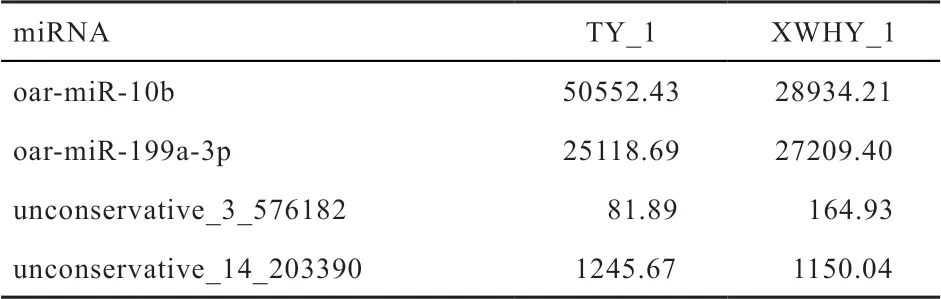
表6 用于qRT-PCR 驗證的miRNA 測序表達量數據
2.8 差異表達miRNA 分析 利用IDEG6 對上述鑒定出的miRNAs 進行表達差異篩選,獲得TY_1 與XWHY_1樣品間差異表達的miRNA,篩選標準為|log2(FC)|Co且FDRg2 為71,其中FC 為差異倍數(Fold Change);FDR 為錯誤發現率(False Discovery Rate)。結果在樣品TY_1 與XWHY_1 共篩選到63 個上調miRNAs 和16個下調miRNAs。進一步對在2 個樣品中差異表達的79個miRNAs 進行分析,發現43 個為已知miRNA,36 個為新發現miRNA。最后對43 個差異表達的已知miRNA進 行 聚 類 分 析( 圖3),oar-miR-127、oar-miR-379-5p、oar-miR-10a、oar-miR-411a-5p 和oar-miR-493-5p等在TY_1 和XWHY_1 樣品中表達量均較高,而oarmiR-376 家族(包括oar-miR-376b、c、d 和e)和oarmiR-3959-3p 在2 個樣品中的表達量差異較顯著(差異倍數大于4),提示其可能在不同品種綿羊皮膚毛囊發育中起重要的調控作用。
2.9 差異表達miRNA 靶基因預測及富集分析 根據篩選到的差異表達miRNA 與綿羊的基因序列信息,用miRanda 和RNAhybrid 軟件進行靶基因預測。使用BLAST 軟件將預測靶基因序列與GO 和KEGG 等數據庫比對,獲得靶基因的注釋信息分別為3 886 和4 449個。最后對樣品TY_1 和XWHY_1 間差異表達miRNA靶基因進行GO 富集分析和KEGG 中通路類型分類統計,GO 統計發現其主要參與代謝過程、催化活性、細胞進程和細胞組分等過程(圖4);KEGG 通路的結果表明,差異表達miRNA 的4 449 個靶基因富集到113個信號通路上,其中在嘌呤代謝、內吞作用和糖酵解/糖異生等信號通路上富集顯著性較高。
3 討 論
皮膚毛囊的生長發育受到多個信號分子及信號通路的協同調控,是一個復雜的分子調控過程[13]。從miRNA 的角度簡析綿羊皮膚毛囊生長發育的機制,對于最終從分子水平上揭示綿羊皮膚毛囊形成的分子機理及其調控機制具有重要作用。
有研究證實,灘羊在1 月齡時可宰取理想的羔皮,而小尾寒羊從胎內到3 或4 月齡都可宰取理想的羔皮[14]。為探討這種差異形成的分子機制,本研究運用高通量測序技術分析比較了TY_1 和XWHY_1 皮膚毛囊組織中的miRNA 表達差異。測序原始數據經過去低質量、去污染和去接頭等一系列處理,在灘羊和小尾寒羊皮膚毛囊組織樣品中分別得到16 350 847 個和17 891 155 個高質量序列讀數。通過與miRBase 數據庫(V21.0)比對,在2 個樣品中共鑒定出561 個miRNA,其中138 個為已知miRNA,423 個為新發現miRNA。通過篩選TY_1 與XWHY_1 間差異表達miRNA 發現63 個為上調miRNAs,16 個為下調miRNAs。這些差異表達miRNA中43 個為已知miRNA,36 個為新發現miRNA。
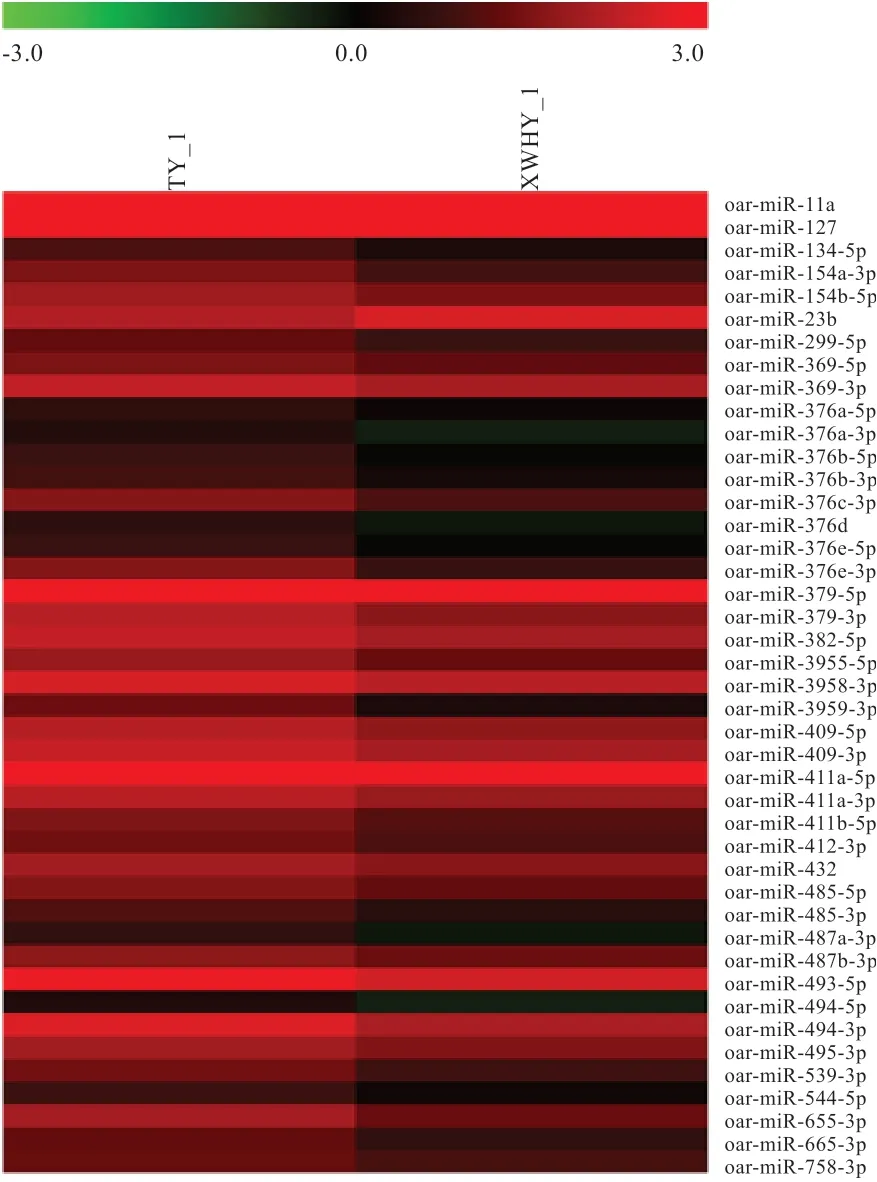
圖3 差異表達的已知綿羊miRNA 聚類分析
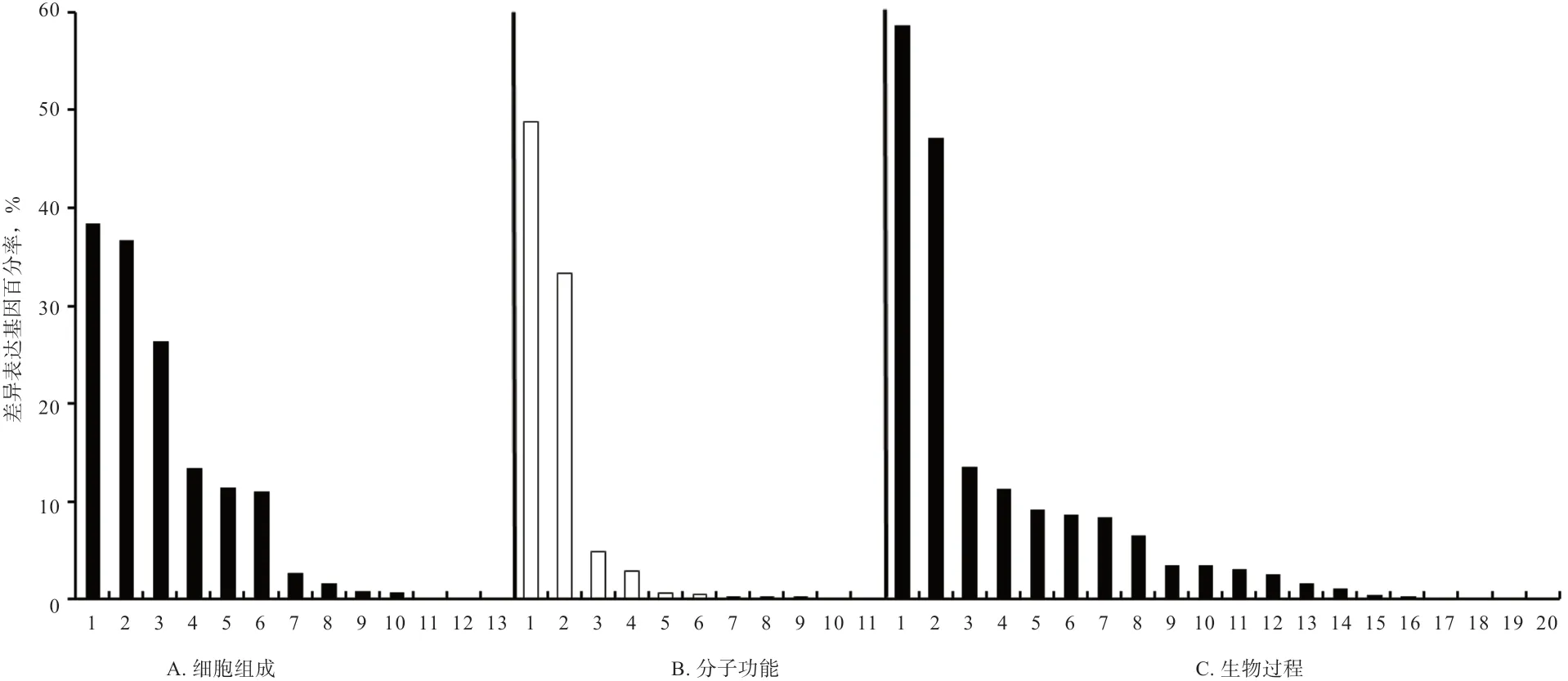
圖4 差異表達miRNA 靶基因的基因本體(GO)富集分析
相對于妊娠45 d 的絨山羊,在妊娠55 d 和65 d 樣本中oar-let-7 和oar-miR-200 家族的miRNA 表達量都顯著上調,提示其可能是毛囊發育中關鍵的miRNA[15]。而在本研究中前20 個高表達的已知miRNA 中oar-let-7 家族有5 個,分別為oar-let-7f、oar-let-7a、oar-let-7g、oarlet-7i 和oar-let-7c;而oar-miR-200 家族有2 個,分別為oar-miR-200b 和oar-miR-200c。 另 外,oar-miR-200 家族在羊駝皮膚和毛囊發育中也可能起重要作用[16]。已有研究報道,oar-miR-30a-5p 和oar-miR-23a 等可通過調控沉默信息調節因子2 相關酶1(Silent mating type information regulation 2 homolog 1,SIRT 1)參與羊毛卷曲生長[17],而在本研究中也檢測到oar-miR- 30a-5p和oar-miR-23a 的高表達,提示其可能參與小尾寒羊和灘羊毛發卷曲生長。另外,本研究在灘羊和小尾寒羊皮膚中檢測到高水平表達的oar-miR-125b 在羊駝背部皮膚中表達量也較高[16]。與小尾寒羊相比,在灘羊皮膚毛囊中高水平表達的oar-miR-127 和oar-miR-379-5p,在藏綿羊皮膚毛囊中也檢測到[18]。而oar-miR-376 家族(包括oar-miR-376b、c、d 和e)和oar-miR-3959-3p在2 個樣品中的表達量差異最顯著(差異倍數大于4),提示其可能在不同品種綿羊皮膚毛囊發育中起重要的調控作用。GO 統計差異表達miRNA 的靶基因發現,其主要參與代謝過程、催化活性、細胞進程和細胞組分等過程;KEGG 通路的結果表明,差異表達miRNA 的靶基因主要富集到嘌呤代謝、內吞作用和糖酵解/糖異生等信號通路上。
4 結 論
本實驗利用高通量測序技術在灘羊和小尾寒羊皮膚毛囊組織中篩選得到了79 個差異表達的miRNAs,并對其靶基因進行預測,推測其可能參與了綿羊皮膚毛囊發育的調控,但其具體機制還需要進一步驗證。
猜你喜歡
英語世界(2023年10期)2023-11-17 09:19:16
汽車實用技術(2022年10期)2022-06-09 11:16:58
音樂探索(2022年2期)2022-05-30 21:01:37
收藏界(2019年3期)2019-10-10 03:16:40
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國特種設備安全(2018年11期)2019-01-08 02:08:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
中國非營利評論(2017年1期)2017-11-09 03:09:10
海外華文教育(2017年8期)2017-11-07 04:42:02
現代語文(2016年21期)2016-05-25 13:13:50
