保健品中原花青素含量檢測方法的比較研究
2019-07-31 06:36:32汪玉玲陳星蓉陸婷婷林騰奕梁培榮
質量安全與檢驗檢測 2019年3期
關鍵詞:方法
汪玉玲 陳星蓉 陸婷婷 李 桑 林騰奕 梁培榮
(廣東省食品檢驗所 廣東廣州 510435)
1 前言
原花青素(Procyanidins)是植物中一大類由兒茶素、表兒茶素、沒食子酸和表兒茶素沒食子酸酯等單體鍵合而成的聚多酚化合物的總稱[1],是一種應用前景非常廣闊的新型植物性抗氧化劑,能強效地清除人體自由基。研究表明[2],原花青素在體內抗氧化、清除自由基能力是維生素C 的20 倍、維生素E的50 倍,能防治由自由基引發的80 多種疾病,有預防心臟病、保護大腦神經系統[3]、降低血脂、抗腫瘤[4]、抗氧化(延緩衰老)[5]、調節免疫力[6]及保護視力等作用。近年來,以葡萄籽為提取原料的原花青素已廣泛用于食品、藥品、保健品及化妝品領域中,我國對含原花青素的保健品也進行了大量的研制開發和應用,市場發展迅猛,但產品質量參差不齊。因此,建立一種準確高效的測定保健品中原花青素含量的方法具有十分重要的意義。
目前,我國對保健品中原花青素的檢驗是用鐵鹽催化比色法。其測定原理是原花青素在加熱的酸性條件且和鐵鹽催化作用下,C-C 鍵斷裂而生成深紅色花青素離子(即氰定),測定在特征波長525 nm處的吸光度。但在實際操作中發現,樣品采用沸水浴回流加熱,操作煩瑣,易受氧氣[7]、日照、溫度等因素的影響,平行性較差,不適于日常大批次的保健品中原花青素的檢測;且比色法分離能力差,不能較好地排除其他有色雜質對待測物的影響,測定結果的準確度較低。因此,對于保健品中原花青素含量的測定是目前面臨的一個難題。
在已有的相關文獻中,對原花青素的檢測方法主要有鐵鹽催化比色法[8]、香草醛-鹽酸法[9]、正丁醇-鹽酸法[10]、高鐵鹽-鐵氰化鉀比色法[11]、紫外分光光度法[12]等,這些方法都要求反應中的特征性顯色明顯,且顯色穩定時間短,不能對相似的雜質成分進行分離測定,較適于測定原花青素純度高的產品,對一般原料中原花青素的測定分離能力還不夠理想,準確度和靈敏度較低。為此,本文在鐵鹽催化比色法的基礎上,優化了前處理過程,結合高效液相色譜法的高選擇性和高準確度,建立了一種適用于分析保健食品中原花青素含量的鐵鹽催化-高效液相色譜法[13,14],該方法前處理較簡便、重現性好,可以有效地分離待測樣中共存的其他有色雜質,避免干擾,有較高的準確度和靈敏度,適于實驗室大批次的檢驗工作,可作為快速測定保健食品中原花青素含量的一種參考方法,為保健品的質量監管提供技術支撐。
2 材料與方法
2.1 材料與試劑
原花青素標準品(純度≥95.2%,上海源葉生物科技有限公司);甲醇(色譜純,美國默克公司);甲酸(色譜級,上海阿拉丁生化科技有限公司);異丙醇(色譜純,美國BCR 公司);二氯甲烷(色譜純,上海麥克林生化科技有限公司);正丁醇(分析純,廣州化學試劑廠);鹽酸(分析純,廣州化學試劑廠);硫酸鐵銨(分析純,廣州化學試劑廠);超純水由實驗室制備。
實驗樣品:原花青素葡萄籽軟膠囊、原花青素葡萄籽精華片、原花青素桑葚黑枸杞果汁口服液(均為市售常見商品)。
2.2 儀器與設備
Prominence UFLCXR 高效液相色譜儀、SPD-M20A檢測器(日本島津公司);UV-2700 紫外/可見分光光度計(日本島津公司);Milli-Q Reference 超純水儀(美國密理博公司);HWS28 型電熱恒溫水浴鍋(上海一恒科學儀器有限公司);電熱鼓風干燥箱(上海一恒科學儀器有限公司);KQ-500DE 型臺式數控超聲波清洗器(昆山市超聲儀器有限公司);MS105DU萬分之一天平、PL602E 電子天平(瑞士梅特勒公司);20 mL 棕色頂空瓶(浙江歐爾賽斯科技有限公司);0.22 μm 針孔濾膜(德國波爾公司)。
2.3 實驗方法
2.3.1 樣品制備及提取
(1)片劑:將試樣研磨成粉狀后,稱取1 g(精確至 0.001 g)于 100 mL 容量瓶中,加入 50 mL 甲醇,超聲處理20 min,放冷至室溫后,加甲醇至刻度,搖勻,離心至澄清后取上清液備用。
(2)膠囊:擠出膠囊內容物,攪拌均勻,稱取50 mg(精確至0.001 g)試樣置于小燒杯中,用少量二氯甲烷使試樣溶解,并洗入200 mL 棕色容量瓶中,加甲醇至刻度,搖勻。
(3)口服液:搖勻后取 1 g(精確至 0.001 g)試樣置于25 mL 棕色容量瓶中,加甲醇至刻度,搖勻。
2.3.2 水解反應
取正丁醇:鹽酸(95:5,V/V)混合溶液 6 mL 置于20 mL 空瓶中,再加入0.2 mL 硫酸鐵銨溶液(稱取2 g硫酸鐵銨,用濃度為2 mol/L 鹽酸溶解,定容至100 mL)和樣品溶液1 mL,加蓋密封,混勻,置100℃烘箱中加熱40 min,然后立即置冰水中冷卻,過0.22 μm 濾膜,備用。
2.3.3 標準曲線制備
原花青素標準品經純度換算后精確稱取0.052 9 g,用甲醇溶解并轉移到50 mL 棕色容量瓶中,定容到刻度,配制成質量濃度為1.00 mg/mL 的標準溶液,現配現用。用移液槍分別準確移取 50 μL、125 μL,250 μL,500 μL,750 μL,1 500 μL 標準溶液于 5 mL棕色容量瓶中,用甲醇至刻度,搖勻。各取1 mL 用以上步驟進行水解,并制備工作曲線。
2.3.4 色譜條件
色譜柱:Agllent ZORBAX SB-C18 StableBond Analytical (4.6 mm×250 mm,5 μm);流動相:水+甲醇+異丙醇+甲酸=65+22+5+8(V/V);流速:1 mL/min;柱溫:30℃;進樣體積:10 μL;檢測波長:525 nm。
3 結果與分析
3.1 加熱方法的優化
利用沸水浴回流加熱40 min 后測定, 但實際操作中發現,沸水浴回流操作煩瑣,較難適用于實驗室大批量的樣品檢測。針對此問題,本實驗將沸水浴回流改進成如下2 種方法:(1)改用25 mL 具塞比色管,于 100°C 沸水浴加熱 40 min 反應;(2)改用 20 mL 的棕色頂空瓶,放入 100°C 烘箱加熱 40 min。具體操作:選用原花青素葡萄籽軟膠囊作為試樣,分為低、中、高3 組,分別加入約50%、100%、200%試樣含量的標準溶液,同時考察沸水浴回流、直接沸水浴加熱及烘箱加熱3 種反應方式下的原花青素含量,重復實驗3 次(n=3)。不同加熱方法的比較結果詳見表1。
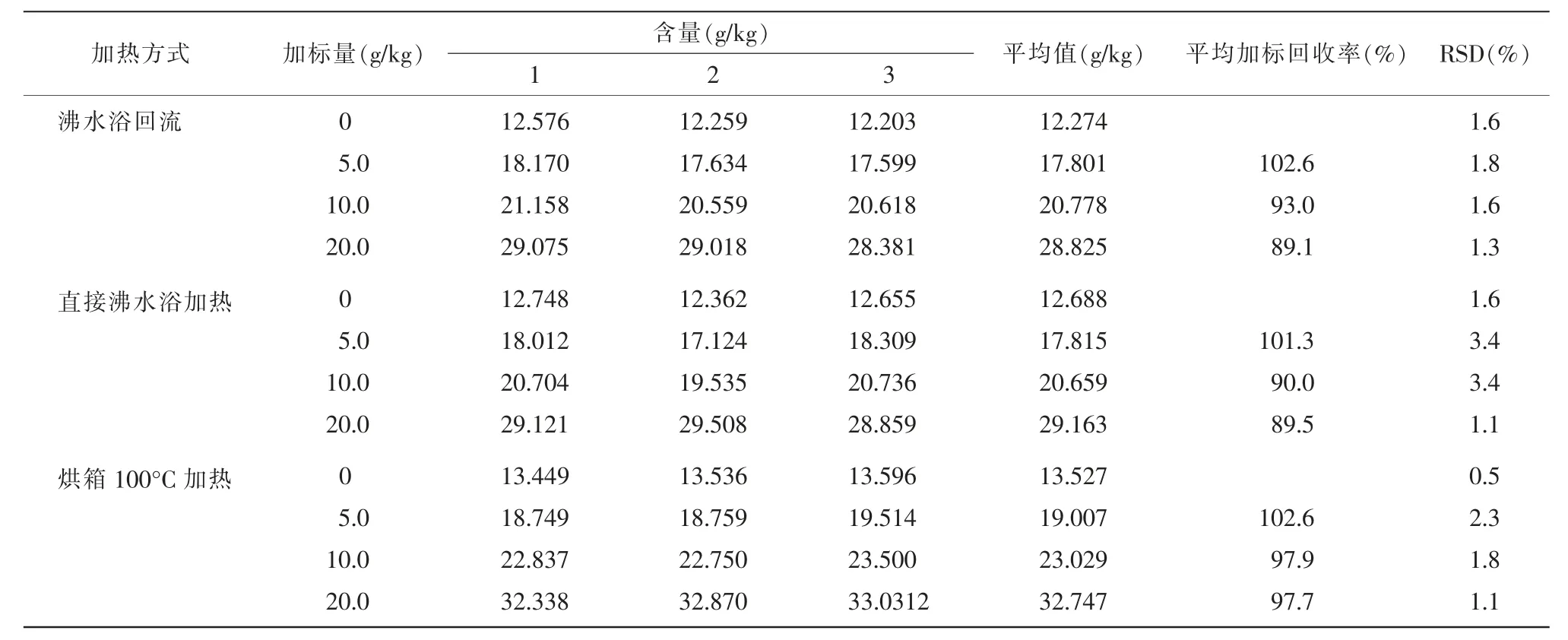
表1 不同加熱方法的比較
由表1可看出,沸水浴回流和直接沸水浴加熱這2 種方式測得的樣品含量相差不大,但中、高濃度組的加標回收率較低,與烘箱加熱相比,后者的試樣測定值明顯高于前兩者,且3 個水平的加標回收率都優于前兩者(97.7%~102.6%),相對標準偏差(0.5%~2.3%)也滿足理化檢驗要求,故采用烘箱100°C 加熱的方式較優。其原因可能與采用密封的棕色頂空瓶和相對密閉的烘箱環境有關,此操作可有效地隔絕反應過程中氧氣、光照對原花青素的氧化,減少實驗過程的損失,提高加標回收率,故將樣品前處理的加熱方法優化為用頂空瓶于烘箱加熱。
3.2 水解反應的溫度選擇
據相關文獻表明[15],原花青素的水解反應需要較高的溫度,室溫下反應速度極慢,在不同水解溫度下,花青素離子的生成量差異很大,且該差異不能通過延長加熱時間來彌補。為探索原花青素最佳的水解溫度,本文將含量為100 mg/L 的原花青素標準溶液置于棕色頂空瓶中密封后在在烘箱中加熱,水解反應 40 min,分別對比在 80℃、90℃、100℃、110℃、120℃下5 種烘箱溫度下原花青素的測定值。詳見圖1。
由圖1可知,當溫度低于100℃時,原花青素的水解反應不完全,隨著溫度的升高,水解反應速度加快,花青素離子的生成量逐漸增加;當溫度在100℃~110℃時,花青素離子的生成量達到最大值;當溫度高于110℃,花青素離子的生成量開始有所下降。
其可能的原因是:不同溫度會影響不同聚合度的原花青素的C-C 鍵的斷裂程度。隨著溫度的升高,原花青素單元間的連接鍵更易斷裂,使得花青素離子的生成量增加;但原花青素極不穩定,在高溫下又極易失去活性,發生自氧化的現象[16],從而使花青素離子的生成量降低。故綜合考慮,本試驗采用100℃作為原花青素的最佳水解溫度。
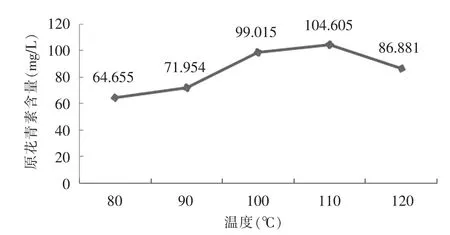
圖1 不同溫度下原花青素含量的變化圖
3.3 水解反應時間的選擇
為了探索最佳的水解反應時間,本試驗將已知含量的原花青素試樣置于棕色頂空瓶中密封后于100℃烘箱中加熱,分別比較加熱反應20 min、40 min、60 min和80 min 時原花青素的測定值。詳見圖2。
由圖2可知,加熱約40 min,測得的試樣中原花青素含量達到最大值;40 min 后,隨著加熱時間的延長,結果測定值稍有下降。可能的原因是:高溫條件下,原花青素發生自氧化現象,隨時間的延長原花青素不斷降解,導致含量不斷減少,故本試驗采取水解反應40 min。
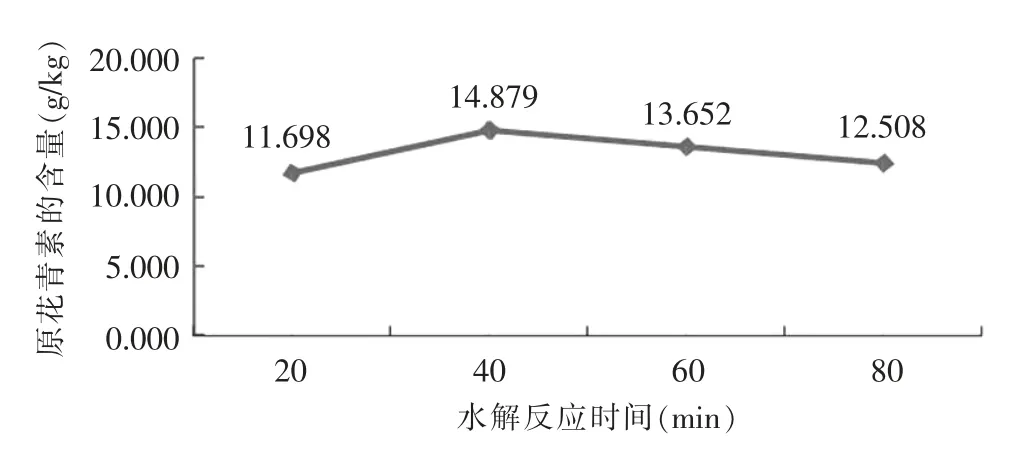
圖2 不同水解反應時間下原花青素含量的變化圖
3.4 硫酸鐵銨催化劑用量的選擇
原花青素在加熱的酸性條件下氧化為花青素離子,是一種氧化還原反應。早期有人提出,Fe3+可能通過與花青素離子絡合作用, 增大吸光系統。后來Lawrence 等[17]證實,在原花青素生成花青素的過程中, Fe3+起到催化劑的作用,可加快反應速率。本文為了考察硫酸鐵銨催化劑的最佳用量,取已知含量的原花青素軟膠囊為試樣,分別對比了在不加、加1%、加2%及加3%硫酸鐵銨溶液4 種情況下測得的試樣中原花青素的含量。結果詳見圖3。
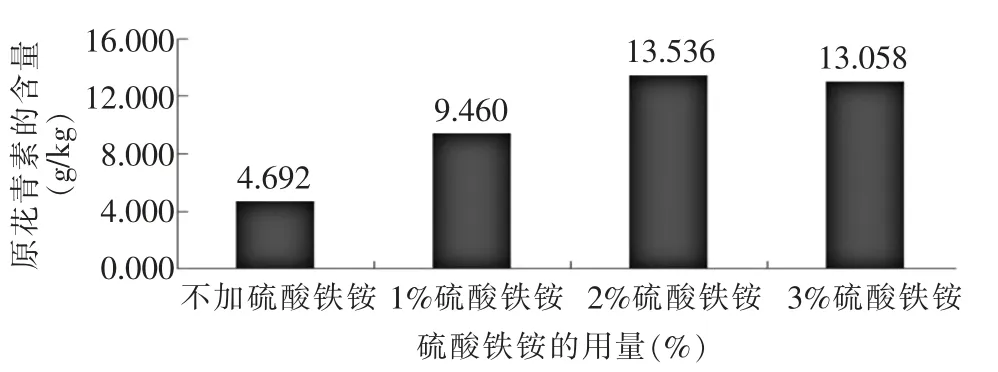
圖3 催化劑用量對原花青素測定的影響
由圖3可知,不加硫酸鐵銨溶液的試樣水解后顏色明顯比加硫酸鐵銨溶液的顏色淺,同一樣品不加硫酸鐵銨溶液時測定值最低,加2%硫酸鐵銨溶液時,測定值最高,之后增加用量,測定值都趨于穩定。故采用2%硫酸鐵銨溶液做催化劑,既節省試劑的用量又可使水解反應更完全。
3.5 檢測方法的比較
3.5.1 色譜法與比色法準確度的比較
目前對原花青素的檢測采用鐵鹽催化比色法。鑒于高效液相色譜法具有較強的分離能力、較好的定性和定量效果,故本試驗嘗試使用高效液相色譜法進行分析(圖4)。比對時采用相同的樣品、試劑和優化后完全一致的前處理方法,對處理好的樣品分別使用鐵鹽催化比色法和色譜法進行分析測定。
比較發現:2 種方法的平行性和加標回收率都滿足要求,但當試樣為含油軟膠囊時,鐵鹽催化比色法測定結果高于色譜法,結果詳見表2。且由圖4可看到,目標峰原花青素的右側有一較小的雜質峰,且該雜質峰在525 nm 波長附近同樣有較大吸收(圖5)。故可能是由于比色法沒有分離能力,當原花青素試樣成分復雜時,部分有色雜質與待測物混在一起干擾測定,從而使比色法的測定結果偏高。故采用高效液相色譜法測定保健品中原花青素的含量結果更為準確。
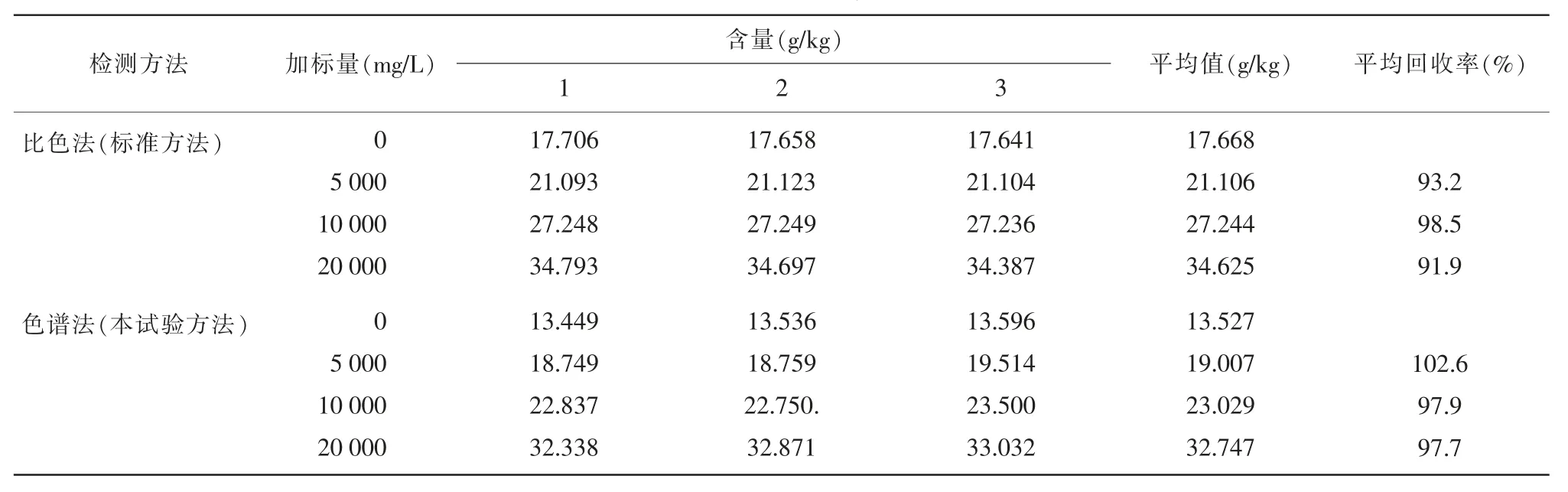
表2 2 種方法測定結果的比較
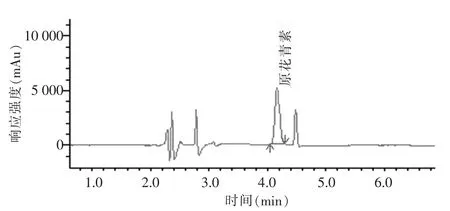
圖4 試樣在色譜法中分離的色譜圖
3.5.2 穩定性試驗的比較
取已知含量的原花青素試樣加入相同的試劑,在相同條件下進行水解反應,對處理好的試樣每隔1 h 分別采用鐵鹽催化比色法和色譜法進行分析測定,觀察樣品測定值隨時間變化的規律,結果詳見圖6。
由圖6(a)、(b)可知,鐵鹽催化比色法在試樣溶液顯色2 h 內測定吸光度值較穩定;而高效液相色譜法在上機的5 h 內,測定結果都基本穩定,含量無顯著變化,(相對標準偏差)RSD 為0.5%。
因此,綜合考慮,本試驗采用高效液相色譜法進行分析測定,該方法相比于鐵鹽催化比色法,檢測結果準確度更高,穩定性更好。
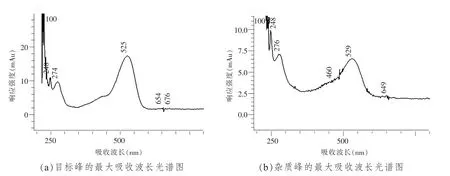
圖5 目標峰和雜質峰的最大吸收波長光譜圖
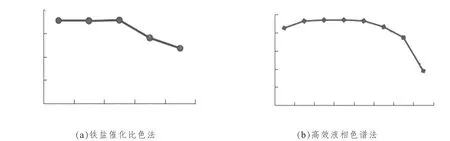
圖6 測定值隨時間的變化趨勢
3.6 方法線性范圍及檢出限、定量限
將原花青素系列工作溶液(10 mg/L、25 mg/L、50 mg/L、100 mg/L、150 mg/L、300 mg/L)按優化后的實驗條件進行水解反應,按上述色譜條件進樣分析,得到原花青素的色譜圖,原花青素標準品的濃度和對應的峰面積詳見表3,兩者的線性關系詳見圖7。
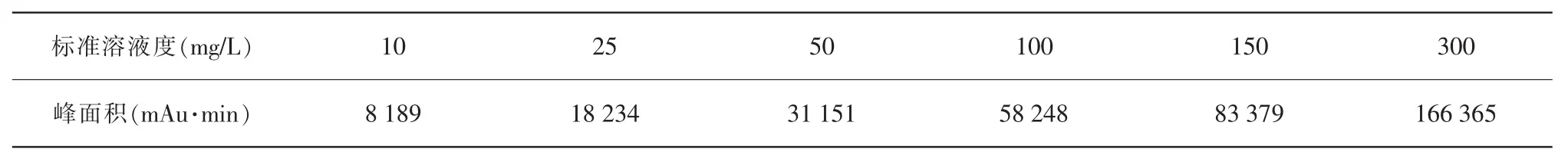
表3 原花青素標準品的濃度及峰面積
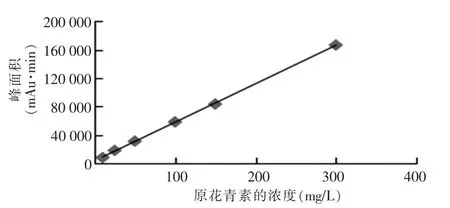
圖7 原花青素的標準曲線圖
由圖7可知,原花青素在濃度為10~300 mg/L時線性良好,線性回歸方程為Y=540.7X+3 694.23,相關系數平方為r2=0.999 7。原花青素峰型良好,保留時間為4.147 min,理論塔板數為9 613。
以已知原花青素含量的試樣為實驗對象,按優化后的方法進行前處理,考察其檢出限,以目標化合物色譜峰3 倍信噪比(S/N)計算方法檢出限(Limit of Detection, LOD),10 倍信噪比計算方法定量限(Limit of Quantization, LOQ),得到原花青素的方法檢出限為1.5 mg/kg,方法定量限為5.0 mg/kg。
3.7 方法的精密度和準確度
分別以原花青素葡萄籽軟膠囊、原花青素葡萄籽精華片、原花青素桑葚黑枸杞果汁口服液為試驗對象,各分為低、中、高3 組,每組分別加入約50%、100%、200%試樣含量的標準溶液,按照本方法優化好的條件進行測定,每組重復試驗6 次(n=6),以加標回收率考察本方法的準確度,以相對標準偏差考察方法的精密度,結果詳見表4。由表4可知,3 種類型保健品中原花青素的加標回收率為99.0%~103.6%,相對標準偏差(RSD)均小于2.2%,符合理化檢驗分析方法要求,故本試驗所建立的分析保健品中原花青素含量的方法具有可靠的準確度和精密度。
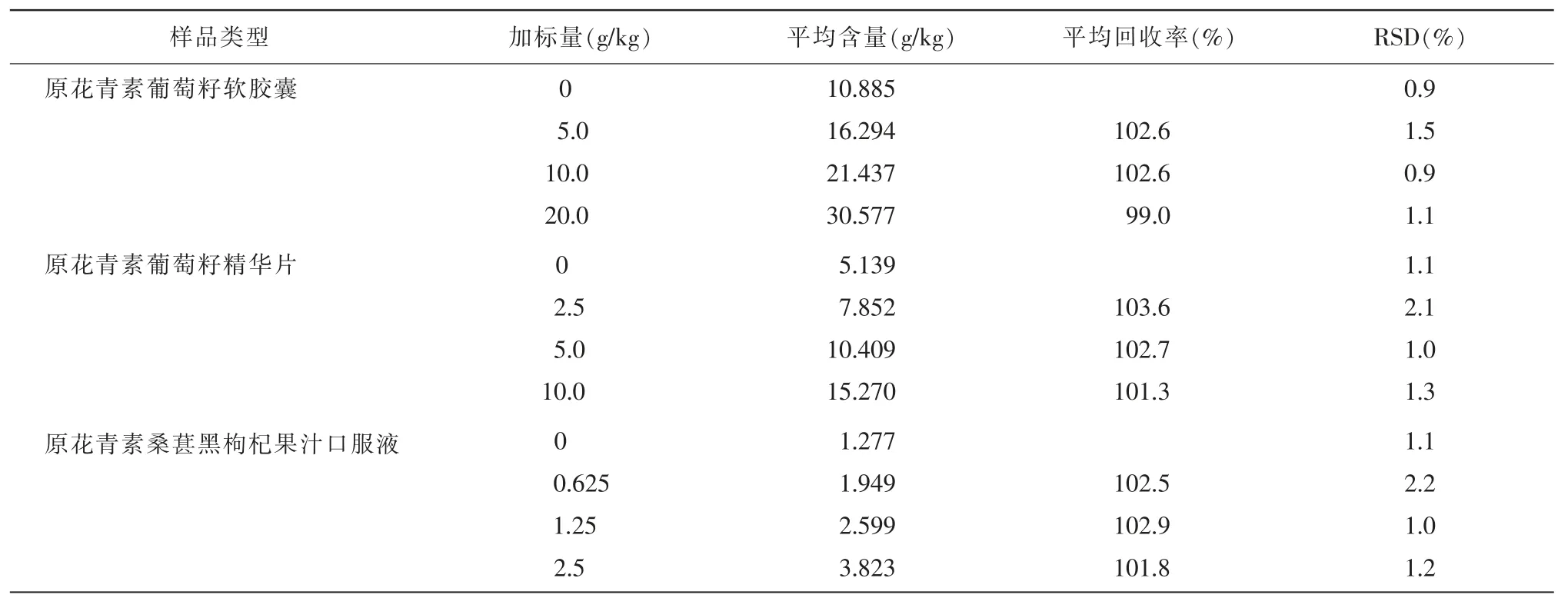
表4 回收率和精密度實驗結果
4 討論
與以前的保健食品中原花青素的測定方法比較,本試驗具有以下優勢:(1)在加熱方式的選擇上,采用頂空瓶于烘箱加熱代替沸水浴回流加熱簡化了煩瑣的樣品前處理過程,適于實驗室大批量樣品的快速檢驗,節約時間,可提高檢驗效率;同時,密閉的烘箱能有效地隔絕氧氣和光照,減少氧化,提高測定的準確性、平行性;(2)樣品前處理上,在鐵鹽催化劑的用量、加熱溫度和水解時間3 方面做了對比研究,結合原花青素的性質,確定了最適原花青素反應的條件,有助于提高測定的準確度,且為今后探索不同資源的原花青素的組分、結構、性質提供了參考價值;(3)在分析方法的選擇上,采用高效液相色譜法代替鐵鹽催化比色法,提高了分離能力,可排除部分有色雜質對待測物的干擾,且靈敏度、精密度、準確度和穩定性都較好,滿足檢驗需求,可作為快速檢測保健食品中原花青素含量的參考方法。
近年來,由于原花青素對人體有多種特殊功效,含原花青素的保健品已不斷被研究開發利用,市場需求旺盛,與此同時也出現相關質量問題,目前我國關于原花青素的測定方法也尚無統一的標準,故本文所優化的快速準確測定保健品中原花青素含量的方法對日常保健品的質量監管有一定的指導意義和應用推廣價值。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
