水蘇堿在乙醇中的納濾分離行為
2019-07-23 11:42:06李存玉李賀敏支興蕾劉莉成李紅陽彭國平
中成藥 2019年6期
李存玉, 李賀敏, 支興蕾, 劉莉成, 李紅陽, 彭國平?
(1.南京中醫藥大學藥學院,江蘇南京210023;2.江蘇省中藥資源產業化過程協同創新中心,江蘇南京210023)
以乙醇為代表的有機溶劑在中藥制藥領域中應用廣泛,但衍生出的提取物、萃取物、洗脫部位在進行精制純化時,高溫處理所帶來的成分轉化會嚴重影響目標產物質量,同時因回收溶劑比例變化而難以循環利用, 造成資源浪費、 環境污染[1]。1993 年高從堦院士首次提出常溫化精制的納濾分離技術[2],其截留分子量在100 ~1 000 Da 之間,分離特點與超濾、微濾有所不同,因其膜材質表面的荷電性,分離過程為電荷、位阻、溶解-擴散效應等多種效應綜合的結果[3-4],但針對有機溶液環境下中藥成分的納濾精制研究尚處于探索階段[5]。
水蘇堿是益母草中主要藥效成分[6],在光照和長時間加熱下均會降解,影響制劑質量的同時也造成藥用資源的浪費,在對其進行色譜分離時制得以乙醇為洗脫液的中間體,大多采取低溫減壓回收來控制成分的降解,但生產效率偏低。本實驗探索納濾技術對乙醇中水蘇堿富集的適用性,并結合膜面溶脹特征闡明該成分的分離規律,為明確有機溶液環境下納濾分離機制提供參考依據。
1 材料
1.1 試藥 復合聚酰胺納濾膜(孔徑150、475、800 Da,南京拓鉒醫藥科技有限公司)。鹽酸水蘇堿對照品(批號110773-201313,中國食品藥品檢定研究院)。益母草提取物(自制,益母草飲片加水提取后色譜分離制備得到, 水蘇堿含有量>70%)。乙腈為色譜純;其他試劑均為分析純。
1.2 儀器 TNZ-1 型納濾分離設備(南京拓鉒醫藥科技有限公司);e2695 高效液相色譜儀(美國Waters 公司);ELSD 6000 檢測器(美國Alltech 公司);MS105 電子天平[十萬分之一,梅特勒-托利多儀器(上海) 有限公司]。
2 方法
2.1 溶液制備
2.1.1 對照品溶液 精密稱取鹽酸水蘇堿對照品3.60 mg,置于5 mL 量瓶中,流動相超聲溶解,定容至刻度,即得(每1 mL 含0.72 mg 該成分)。
2.1.2 供試品溶液 益母草提取物用不同體積分數乙醇配制成5.0%、17.5%、30.0%,其中鹽酸水蘇堿質量濃度為105.0 μg/mL。
2.2 納濾分離 采用前期建立的方法[7],按照圖1 組裝納濾膜、中壓泵等納濾裝置,設置操作壓力為1.0 MPa。將納濾膜置于分離系統中,納濾分離初始階段需要藥液在納濾膜中循環平衡,排除因膜材質對成分吸附的干擾,待鹽酸水蘇堿與膜組件之間的吸附、解吸附達到平衡時,從儲液罐中取樣平衡液,進而開始納濾,納濾液罐收集納濾液,待樣品納濾分離至無法維持系統分離時,關閉納濾系統,納濾液中取樣納濾液。
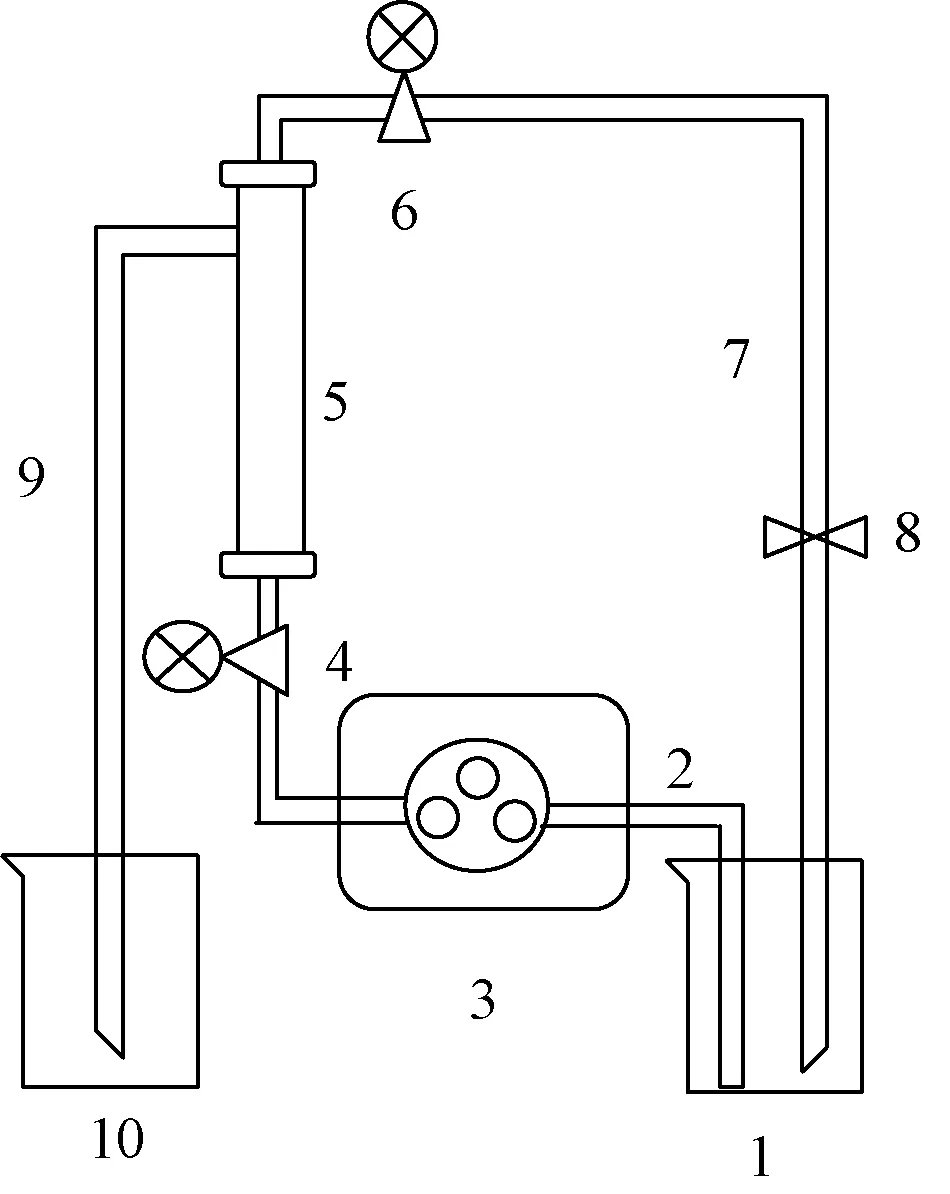
圖1 納濾分離設備Fig.1 Nanofiltration separation equipment
2.3 鹽酸水蘇堿含有量測定
2.3.1 色譜條件[8]Waters Xbridge Hilic 色譜柱(4.6 mm×150 mm,3.5 μm);流動相乙腈-0.2%冰醋酸(80 ∶20)。蒸發光散射檢測器參數為漂移管溫度80 ℃,氣體體積流量2.0 L/min;體積流量0.8 mL/min。
2.3.2 線性關系考察 精密吸取對照品溶液1、2、5、10、20 μL,在“2.3.1” 項色譜條件下進樣測定。以峰面積(Y)、進樣量(X) 取對數進行回歸,得方程為Y=0.56X+4.60(R2=0.999 5),在0.72~14.4 μg 范圍內線性關系良好。
2.4 截留率計算 取納濾分離制備的納濾液(C1) 及平衡液(C2),測定峰面積并取對數后代入“2.3.2” 項下回歸方程,計算鹽酸水蘇堿含有量,按照式(1) 計算截留率(R)。

2.5 水蘇堿分離數據積累 應用Design-Expert 8.06軟件[9],考察乙醇體積分數(A)、pH 值(B)、膜孔徑(C) 對鹽酸水蘇堿納濾分離行為的影響,以截留率(Y)為評價指標。因素水平見表1。

表1 因素水平Tab.1 Factors and levels
2.6 納濾分離層溶脹分析 為了排除膜污染帶來的干擾,在操作壓力1.0 MPa 條件下分別選擇空白水溶液及5%、7.5%、30%乙醇,考察膜面因溶脹帶來的通量衰減變化,見式(2)、 (3)。其中,J0為初始通量,單位m/s;Jt為單位時間t 時的膜通量,單位m/s;m 為膜通量衰減系數。
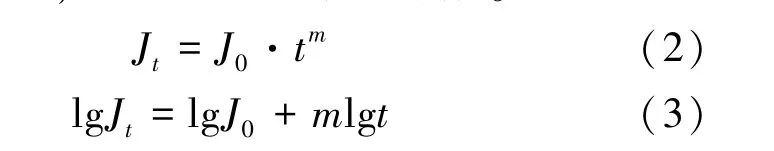
3 結果
3.1 納濾分離數據積累 根據響應面分析原理[10-11],為保障實驗結果的準確性和穩定性,每組平行3 次,取平均值,結果見表2。
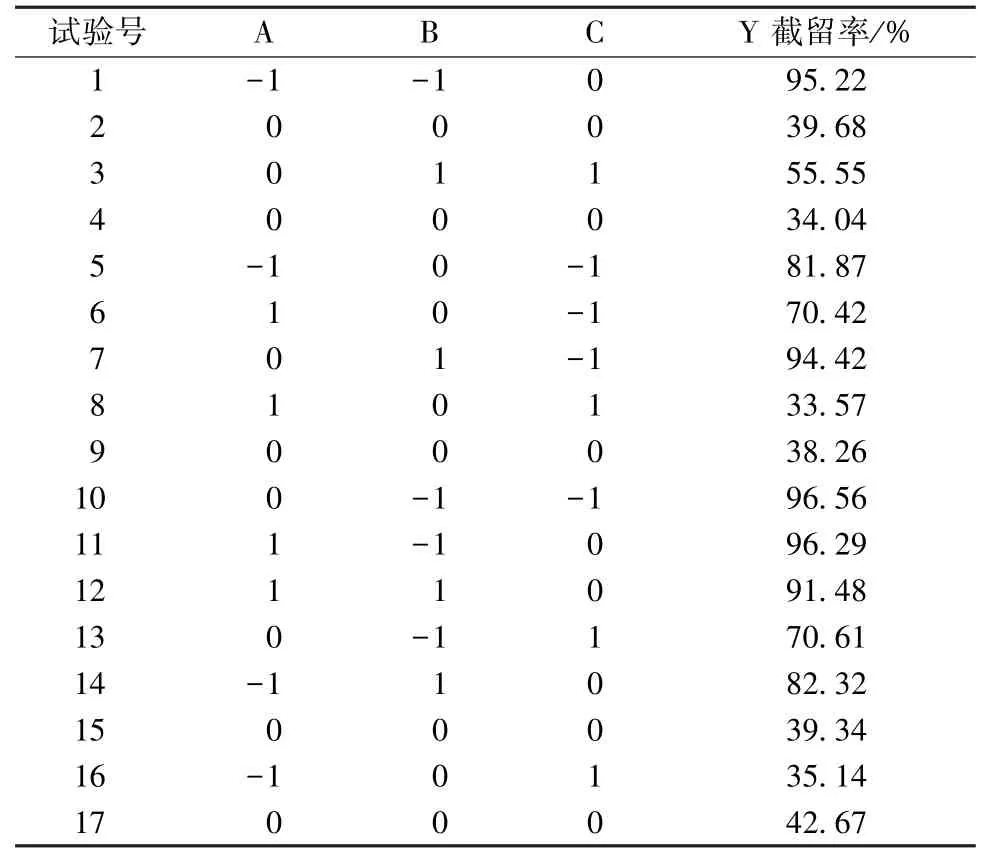
表2 試驗設計及結果Tab.2 Design and results of tests
3.2 模型擬合 對各因素進行二次多項回歸擬合,得方程為Y=39.44-7.26A-4.77B-19.04C+2.43AB+2.96AC-3.23BC+20.52A2+38.28B2+2.20C2,方差分析見表3。由表可知,各因素影響程度依次為C>A>B;P<0.000 1,表明模型極顯著;多元相關系數R2=0.985 7,預測R2=0.824 8,調整R2=0.967 3,表明模型擬合度良好。

表3 方差分析Tab.3 Analysis of variance
3.3 因素分析 圖2 顯示,在固定膜孔徑、乙醇體積分數時,隨著pH 值逐漸升高,截留率呈先下降后升高的趨勢,并在30%乙醇中最明顯,推測水蘇堿兩性結構中堿性、酸性基團解離狀態不同,pH 增加時堿性基團陽離子首先過渡至化合物等電點,然后在堿性環境中羧基解離成陰離子,再結合納濾分離的電荷效應[12];在固定pH 值、乙醇體積分數時,截留率隨著孔徑增加而降低,分離規律符合孔徑篩分規律;在固定pH 值、膜孔徑時,乙醇體積分數對截留率的影響呈不同的調節規律,在酸堿極端環境下,乙醇可增強納濾膜截留效率,但在中性至弱堿條件下,反而會促進成分納濾透過。
表2、圖2 顯示,水蘇堿中的羧基在強堿性條件下以鈉鹽形式存在,與納濾膜電荷排斥效應明顯,在475 Da 納濾中的截留率高于80%,乙醇體積分數從5%升至30%時,截留率由82.32%升至91.48%;在酸性條件下,水蘇堿以鹽酸鹽形式存在,乙醇體積分數從5%升至30%時,截留率由95.22%升至96.29%,推測堿性條件下該成分可能未完全解離,納濾分離時產生的電荷排斥效應弱于酸性條件。
3.4 膜溶脹分析 表4 顯示,隨著乙醇體積分數增加,衰減系數絕對值也隨之升高,相較于水溶液環境,乙醇體積分數增加至5%時單位時間內膜通量衰減明顯,隨著其升高通量衰減加劇,但超過17.5%后衰減系數相對穩定,表明膜溶脹程度已趨于穩定[13]。

圖2 各因素響應面圖Fig.2 Response surface plots for various factors
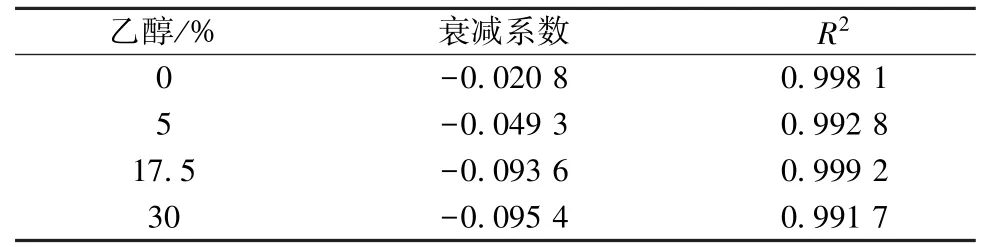
表4 不同乙醇體積分數下納濾膜衰減系數Tab.4 Decay coefficients of nanofiltration membranes under different ethanol concentrations
4 討論
隨著溶液環境由水逐步向有機相過渡,成分狀態、膜面分離層物化性質、成分的跨膜傳質行為均發生變化,進而引起分離行為變化[14-15]。其中,在酸、堿性條件下乙醇體積分數增加引起的水蘇堿截留率升高,表現出不同程度“強化” 截留率的行為,而且其升高程度并不完全是由膜面溶脹引起的。同時表2 顯示,在150 Da 納濾膜中,pH 7.0下乙醇體積分數從5%升高至30%時,截留率由81.87%降低至70.42%,即乙醇體積分數升高引起膜溶脹加劇,水蘇堿截留率反而下降,與上述結果相悖,推測兩性生物堿納濾跨膜傳質機制可能與成分存在狀態相關。
納濾分離的經典模型大多基于水溶液環境建立,其適用性有待于驗證,本實驗中水蘇堿納濾截留行為隨著乙醇體積分數、pH 值變化的現象僅靠電荷效應、位阻效應等分離模型無法得到有效解釋。對水蘇堿存在狀態進行分析,發現隨著溶液pH 由酸性向堿性環境的過渡,該成分呈現出酸、堿性基團解離-游離的轉換狀態,當兩性基團正負電荷數值相等時,即為等電點。由此推測,酸、堿性條件下水蘇堿在乙醇中的解離基團隨著溶劑體積分數升高,其溶解度逐步降低,而游離態基團溶解度變化隨之相反,從而表現出類表面活性劑性質,并通過分子間締合團聚的形式在溶液中存在,故在納濾過濾時“強化” 截留率是由膜面溶脹和分子團聚綜合引起的。另外,在水蘇堿等電點附近呈電中性狀態,由于乙醇可降低分子間表面能,促進多分子團聚態狀態向低分子團聚態或單分子態轉移,而且此時單分子、低分子團聚態水蘇堿比例高于水溶液環境,故此時“削弱” 截留率的行為可能是由存在狀態結構大小、膜面溶脹引起的。
對水蘇堿跨膜傳質過程進行分析,推測在含有乙醇的水溶液環境中,等電點時水蘇堿可能更容易接近膜分離層而通過納濾膜,同時因分子間作用力差異,乙醇與復合聚酰胺材質作用力大于水分子,推測納濾膜表面乙醇體積分數可能稍高于溶液內部。因此,解離態水蘇堿因溶解度與乙醇體積分數呈反比,并在膜面電荷排斥的協同作用下難以進入膜分離層,從而呈現出高截留率現象。
有機溶劑的合理回收和循環利用有助于中藥制藥企業實現節能減排,但分離機制有待于進一步完善。本實驗對兩性化合物水蘇堿在乙醇中的納濾分離行為進行分析,今后將結合分離規律建立納濾傳質模型,從而為提升化學成分在有機溶液環境中的適用性、研制耐有機溶劑納濾膜提供技術支撐。
