基于HPLC聯合ICP-MS法測定活血止痛膠囊中總砷、可溶性砷、價態砷
2019-06-27 05:43:08陳榮張華鋒
中國合理用藥探索 2019年5期
陳榮,張華鋒
(蘇州市藥品檢驗檢測研究中心,江蘇 蘇州 215104)
活血止痛膠囊組方為當歸、三七、醋乳香、冰片、 土鱉蟲、鍛自然銅等六味,收錄于2015年版《中國藥典》一部,功能為活血散瘀,消腫止痛,用于跌打損傷,瘀血腫痛。《中國藥典》[1]對此藥含鉛、砷、汞量作出規定,其中砷含量不得過300 mg/kg,遠高于一般藥物的2 mg/kg。有研究表明[2],當歸藥材對砷元素有明顯吸附作用,對不同產地三七樣品進行分析,砷超標率為32.4%[3],土鱉蟲由于人為增重,食物團比蟲體含砷量明顯提高[4],自然銅作為一種礦物藥,成因復雜,往往含有較高的重金屬及有害元素。因此,評價活血止痛膠囊安全性必須要考慮砷含量問題。而不同砷價態,毒性差別很大。毒性一般為無機砷>有機砷,亞砷酸(三價砷)>砷酸鹽(五價砷)>一甲基砷、二甲基砷,砷甜菜堿和砷膽堿無毒[5]。本實驗采用電感耦合等離子體質譜(ICP-MS)測定活血止痛膠囊中總砷及可溶性砷含量,再以高效液相色譜-電感耦合等離子體質譜(HPLC-ICP-MS)法測定價態砷的含量,以期為科學評價活血止痛膠囊安全性提供參考。
1 儀器、試劑與材料
1.1 儀器與試劑
儀器:7900電感耦合等離子體質譜儀、Agilent 1260高效液相色譜儀(美國Agilent 公司);CEM MARS 6 CLASSIC 微波消解儀(美國培安公司);Millipore Synergy超純水機(美國Millipore公司);SK5200 HP超聲波清洗器(上海科島超聲儀器有限公司,53 kHz,200 W);Thermo Fisher Multifuge X3離心機(美國Thermo Fisher科技公司)。
試劑:亞砷酸(AsⅢ,1704)、砷酸鹽(AsV,1704)、一甲基砷(MMA,1707)、二甲基砷(DMA,1704)、砷甜菜堿(AsB,1607)和砷膽堿(AsC,1607)溶液標準物質均購于中國科學計量研究院;砷單元素標準溶液(174049-3)購于國家有色金屬及電子材料分析測試中心,濃度為1 000 μg/mL;內標溶液(含Bi、Ge、In、Li、Lu、Rh、Sc、Tb 各 100 μg/mL)和調諧液(含 Ce、Co、Li、Mg、TI、Y 各 1 μg/L)均購于美國Agilent 公司。胃蛋白酶和胰蛋白酶均為生化試劑,購自國藥集團化學試劑有限公司,酶活力(U/g)分別為≥1 200和≥50 000。實驗用水為Millpore超純水機所產超純水,其他試劑均為優級純。
1.2 材料
本實驗共收集7批樣品,其中A廠3批(16070119、16080118、17020128,編號1、2、3);B廠3批(20161208、20161011、20161209,編號 4、5、6);C 廠 1 批(160915,編號 7)。
2 方法與結果
2.1 總砷量和可溶性砷含量測定
2.1.1 ICP-MS工作條件以高純氬為工作氣和載氣,氦氣為反應氣;采用碰撞反應池模式;射頻功率1550 W;載氣流量1.09 L/min;采樣深度10.0 mm;霧化室溫度2℃;數據采集重復次數3,積分時間1 S;蠕動泵轉速0.1 rps。
2.1.2 對照品溶液配制精密量取砷單元素標準溶液適量,用2%硝酸稀釋至2 μg/mL,作為砷單元素貯備液,再以2%硝酸稀釋成0、1、5、10、50 ng/mL的系列標準溶液。
2.1.3 供試品溶液制備
2.1.3.1 總砷量供試品制備取供試品10粒,將內容物研細混勻,取供試品約0.4 g,精密稱定,加硝酸8 mL,置加熱板上90℃預消解30分鐘,冷卻后用微波消解儀消解。消解程序:1000 W,爬坡5分鐘,120℃保持10分鐘;1400 W,爬坡10分鐘,185℃保持25分鐘。取出,將消解液轉入50 mL塑料量瓶中,用水洗滌3次,洗液合并于量瓶中,用水稀釋至刻度,搖勻。離心(3 000 r/min,5 min),精密量取上清液1 mL,置50 mL塑料量瓶中,用水稀釋至刻度,即得。同法制備空白溶液。
2.1.3.2 可溶性砷供試品制備取供試品10粒,將內容物研細混勻,取供試品約0.3 g各兩份,精密稱定,置50 mL塑料量瓶中,分別加入人工胃液和人工腸液至刻度,并稱定重量。超聲使供試品徹底溶散,再置于37℃水浴中超聲2 h(53 kHz,200 W)并不時震蕩搖勻。冷卻過夜(>24 h),精密量取中層液體10 mL置離心管中,離心(3 000 r/min,5 min),精密量取上清液5 mL,至10 mL塑料量瓶中,加0.02 mol/L EDTA-2Na至刻度,精密量取1 mL至頂空進樣瓶中,加2 mL硝酸和0.5 mL鹽酸,密閉,90℃烘箱中反應1 h,冷卻后用水洗入10 mL塑料量瓶中,并稀釋至刻度,即得。同法制備空白溶液。
2.1.4 方法學考察
2.1.4.1 線性關系考察按測定條件以對照品質量濃度為橫坐標(X),對照品峰強度為縱坐標(Y),繪制標準曲線。
Y= 0.0017X+ 1.6811×10-5(R= 1.0000),線性范圍1~50 ng/mL。
2.1.4.2 重復性試驗將樣品1(16070119)分別按照2.1.3.1、2.1.3.2方法各制備6份,測得總砷量RSD(n=6)為7.8%,測得可溶性砷含量RSD(n=6,人工胃液)11.5%和RSD(n=6,人工腸液)為10.8%,說明該方法重復性良好。
2.1.4.3 加樣回收試驗稱取樣品5(20161011)約0.2 g,精密稱定,在測定總砷量時精密加入砷單元素標準 溶 液(1 000 μg/mL)40、60、80 μL( 各 3份 ),按照2.2.3.1項下方法制備,測得平均加樣回收率為98.4%,RSD(n=9)為9.4%;在測定可溶性砷含量時精密加入砷單元素貯備液(2 μg/mL)200、250、300 μL(各3份),按照2.2.3.2項下方法制備,測得人工胃液處理的平均加樣回收率為89.2%,RSD(n=9)為11.7%,測得人工腸液處理的平均加樣回收率為90.8%,RSD(n=9)為7.2%。
2.1.4.4 檢出限標樣空白溶液連續測定11次,以信號響應值的標準偏差(δ)的3 倍所對應的質量濃度作為檢出限,測定砷的檢出限為0.01 ng/g。
2.1.5 總砷量和可溶性砷含量測定7批樣品的可溶性砷含量低于總砷量,占總砷比例較低。見表1。
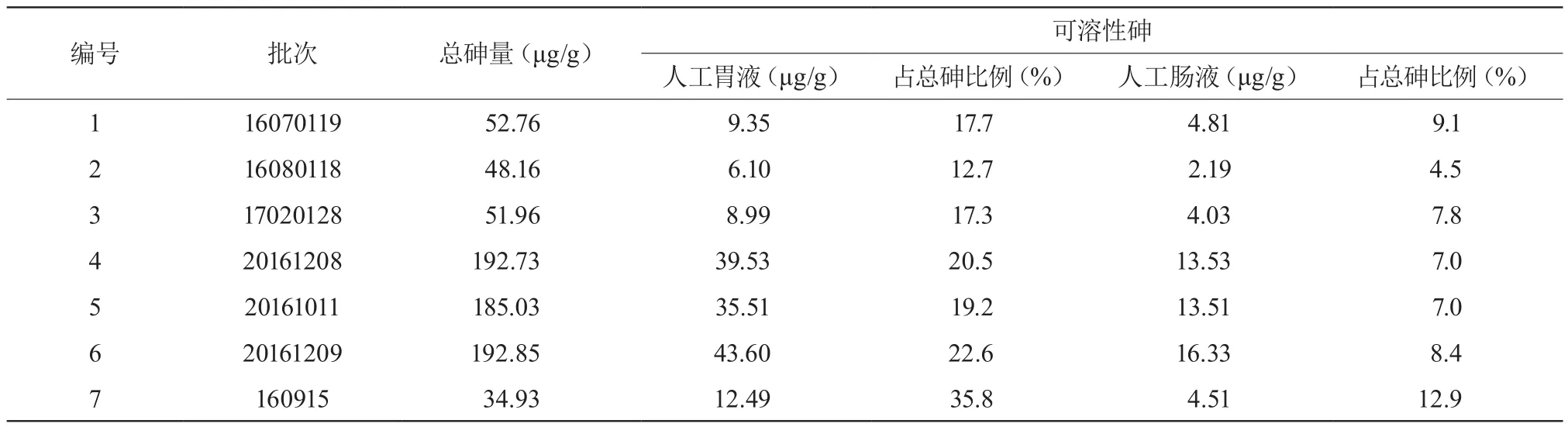
表1 砷含有量測定結果
2.2 價態砷含量測定
2.2.1 ICP-MS工作條件和HPLC色譜條件 ICP-MS工作條件、參數設置與2.1.1基本相同,載氣流量1.10 L/min,蠕動泵轉速0.4 rps。色譜柱:Hamilton PRP-X100(250×4.1 mm,10 μm),0.025 mol/L 磷酸二氫銨溶液(氨水調節pH至8.0)為流動相A,水為流動相B,梯度洗脫(0~15 min,流動相A從0→100%;15~20 min,流動相A從100%→0;20~25 min,流動相A為0),流速為1.0 mL/min,進樣量為20 μL。
2.2.2 對照品溶液制備精密稱取亞砷酸、砷酸鹽、一甲基砷、二甲基砷、砷甜菜堿和砷膽堿適量(以砷As計),加水制成各含2 μg/mL的混合標準貯備溶液,再加 0.02 mol/L EDTA-2Na制成 0,5,20,50,100,500 ng/mL的系列混合標準溶液。
2.2.3 供試品溶液制備同2.1.3.2項下方法制備,并同法制備空白。
2.2.4 方法學考察
2.2.4.1 線性關系考察按測定條件以各價態砷質量濃度為橫坐標(X),以及相應峰強度為縱坐標(Y),繪制標準曲線,各價態砷在相應范圍內呈良好線性關系,見圖1及表2。
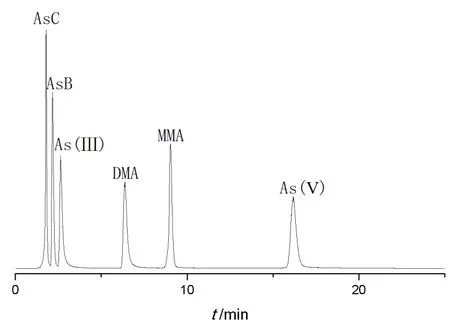
圖1 6 種價態砷 HPLC-ICP-MS 色譜圖
2.2.4.2 重復性試驗7批樣品都只檢出亞砷酸(AsⅢ),將樣品1(16070119)分別按照2.2.3方法制備6份,測得亞砷酸根含量RSD(n=6,人工胃液)為7.5%和RSD(n=6,人工腸液)為11.0%,說明該方法重復性良好。

表2 各價態砷線性關系
2.2.4.3 加樣回收試驗稱取樣品5(20161011)約0.2 g,精密稱定,分別加入亞砷酸根溶液標準物質0.04、0.06、0.08 g(以砷 As計,濃度為 75.7 μg/g),精密稱定各 3 份,按照2.2.3項下方法制備,測得人工胃液處理的平均加樣回收率為91.9%,RSD(n=9)為6.3%,測得人工腸液處理的平均加樣回收率為88.4%,RSD(n=9)為9.2%。
2.2.4.4 檢出限以信噪比(S/N)為3∶1時所對應的濃度為檢出限,計算得到6種價態砷的檢出限分別為:砷膽堿0.19 ng/g;砷甜菜堿0.25 ng/g;亞砷酸根0.35 ng/g;二甲基砷0.50 ng/g;一甲基砷1.02 ng/g;砷酸根0.04 ng/g
2.2.5 樣品中砷價態的測定7批樣品三價砷含量測定結果見圖2及表3。
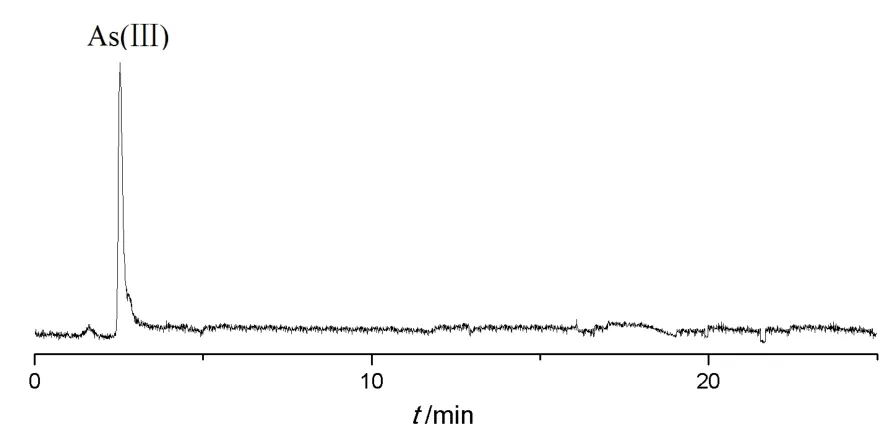
圖2 樣品HPLC-ICP-MS 色譜圖
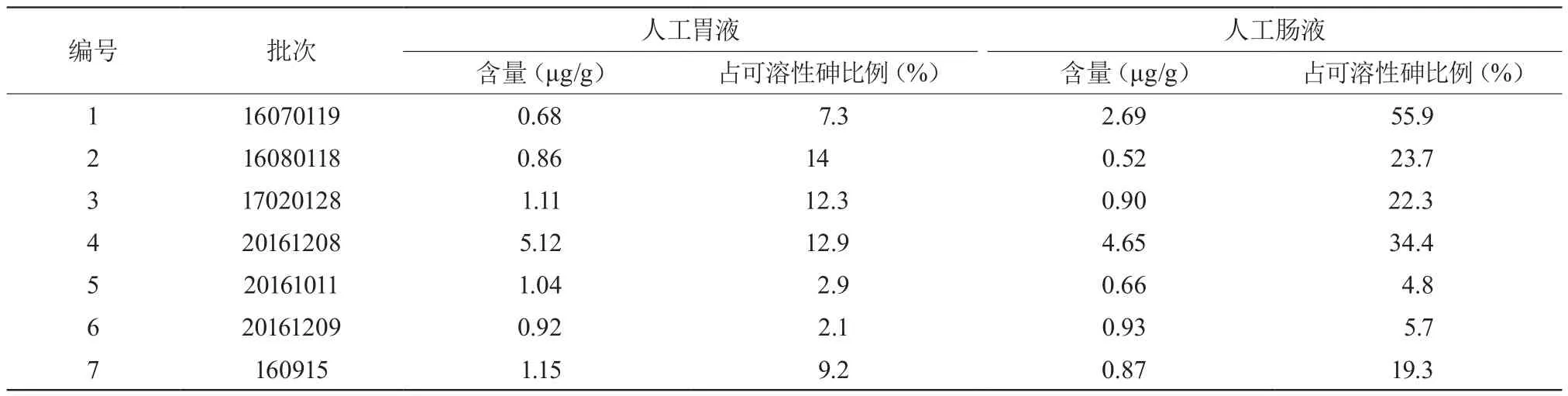
表3 樣品中三價砷含量測定結果
3 討論
含重金屬中藥的毒性與所含重金屬元素價態、形態密切相關,僅采用重金屬總含量來判定藥物的安全性并不合理。特別是中藥復方制劑配伍,及中藥中重金屬在體內吸收、代謝變化規律都有待進一步研究。
活血止痛膠囊是含礦物藥自然銅的復方制劑,生產前后存在砷污染的高風險。本實驗采用微波消解技術提取總砷,同時模擬人體胃腸道環境,分別以人工胃液、腸液提取可溶性砷、價態砷,結合HPLC-ICPMS技術進行測定,結果顯示①活血止痛膠囊可溶性砷及價態砷均遠低于總砷含量,按照體重60 Kg每日最大攝入量3 g計算,可溶性砷攝入量也遠低于WHO規定的0.12 mg[6]。②3家廠商測得總砷量相差較大,但同一家不同批次間相差不多;③7批樣品經人工胃液和腸液分別提取后測得的可溶性砷含量,人工胃液>人工腸液,各批亞砷酸占可溶性砷的比例,人工腸液>人工胃液。多數含有砷的礦物藥是以As(Ⅲ)、As(V)的形式存在,其中As(Ⅲ)毒性更高(約是五價砷的60倍),是造成急性砷中毒的主要兇手[7]。而重金屬及有害元素的生物活性只有在被吸收后才能表現出來,因此在活血止痛膠囊中控制可溶性砷及三價砷含量比單純控制總砷量更為重要。
活血止痛膠囊雖然存在總砷量較高的風險,但在人體內溶出量有限,價態砷含量很低,這可能是因為:①方中采用鍛自然銅入藥,自然銅是硫化物類礦物黃鐵礦(FeS2),晶型多呈立方體,集合體呈致密塊狀[7],經鍛淬后,有害元素大大下降,隨著炮制溫度升高,砷、鉛含量均下降[8],主成分FeS2化學性質穩定,難溶于水,微溶于酸,消解困難,在人胃腸液作用下,砷很難溶出;②本實驗只檢出亞砷酸,且含量與可溶性砷量并不成正比,可能方中砷以實驗以外其他價態存在;③方中其他藥材如果在源頭控制住質量,砷超標的風險就會降低,價態砷也不會超標。
綜上所述,控制含砷風險高的中成藥質量,測定可溶性砷及砷價態含量比單純制定總砷標準更為科學合理。本實驗采用ICP-MS法測定活血止痛膠囊總砷量及可溶性砷含量,同時采用HPLC-ICP-MS測定本品價態砷含量,為活血止痛膠囊的安全性評價提供參考。
