Bradford定量法在蛋白質組學中應用的優化研究
2019-05-28 03:42:36崔為同薛華儒成洪達張海濱王清路
生物技術進展 2019年3期
關鍵詞:標準
崔為同, 薛華儒, 成洪達, 張海濱, 王清路
齊魯醫藥學院, 山東省生物醫學工程技術重點實驗室, 山東 淄博 255300
隨著分子生物學的快速發展,蛋白質組學已成為生物學和生物醫學研究的熱點[1,2]。雙向電泳(2-DE)技術可根據蛋白質的等電點和分子量同時分離多達幾千種蛋白質,因此被廣泛應用于比較蛋白質組學研究[3]。為保證分析的公正和可靠性,等電聚焦電泳時配對分析的兩組蛋白質樣品的加樣量應嚴格一致。這就要求在電泳之前需要對不同樣品進行準確定量。定量準確性不但影響后續蛋白質圖譜分析,而且關系到實驗結果的重現性[4~6]。
為使蛋白質變性并提高蛋白質的溶解度,溶解蛋白質樣品的裂解液(lysis)中含有高濃度的變性劑、表面活性劑、還原劑和兩性電解質等成分,這些成分對常用蛋白質定量方法都存在干擾[7]。已有研究者關注lysis干擾蛋白質定量的問題,但尚未發現完全兼容lysis的定量方法[7~9]。Lysis干擾導致蛋白質定量不準確可能是現有文獻中使用相同實驗材料和同規格膠條,但加樣量存在較大差異的主要原因[10,11]。已有研究分析了Bradford法[12]、二喹啉甲酸(BCA)法[13]和Folin-酚(Lowry)法[14]等三種常用定量方法對2-DE樣品的適用性,發現lysis組分對三種方法都有干擾,但Bradford法對lysis的兼容性最好[7,9]。
Bradford法具有成本低、靈敏度高和測定快速簡便的特點,是目前應用最廣泛的蛋白質定量方法之一[15,16]。雖然該法在測定2-DE樣品時較其他定量方法有一定優勢[7],但仍存在lysis會產生背景干擾的問題[17],而且lysis可能會影響蛋白質-染料復合物的穩定性,但目前尚無相關報道。另外,Bradford在首次報道該方法時沒有明確闡述該法的線性范圍[12],現有文獻資料中對Bradford法線性范圍的描述也不一致[18,19]。
為使Bradford法更適合在蛋白質組學研究中應用,本研究針對目前應用該法定量2-DE樣品時存在的問題,對該法的線性范圍、lysis組分、測定時間、顯色液中磷酸含量、標準曲線的制作以及加樣體積等進行了優化。這些優化可使Bradford法更適合定量蛋白質組學樣品,從而獲得更準確的定量結果。同時可規范定量操作,盡量減小不同實驗者之間定量數據的差異性,使定量數據有較高的參考價值。
1 材料與方法
1.1 材料
1.1.1藥品和試劑 牛血清白蛋白(BSA)(購自Roche公司)、尿素和硫脲(購自Sigma公司)、兩性電解質Pharmalyte (pH 3~10)(購自GE Healthcare公司)、考馬斯亮藍G-250(購自Amresco公司)、二硫蘇糖醇(DTT)(購自Merck公司)、3-[3-(膽酰胺丙基)二甲氨基]丙磺酸內鹽(CHAPS)(購自Promega公司)、乙基苯基聚乙二醇(NP-40)(購自Fluka公司)和聚乙二醇單辛基苯基醚(Triton X-100)(購自Amresco公司),所用藥品均為分析純。試劑配制用水均為蒸餾水。
以BSA為標準蛋白質配制標準蛋白溶液。標準蛋白溶液濃度用比色法校正,即用紫外分光光度計和1 cm光徑的石英比色杯測定1 mg/mL BSA溶液時,在280nm處的吸光度應為0.66[20]。該標準液分裝后保存于-20℃的冰箱中。
顯色液參考Bradford法[12],略作改動。具體為稱取100 mg Coomassie Brilliant Blue G-250,加入50 mL 95%乙醇中。CBB-G250充分溶解后,再向其中加入120 mL 85%(w/V)的磷酸[21],混勻后用蒸餾水定容至1 L并過濾。濾液用棕色瓶室溫保存。
本研究采用常規的尿素/硫脲裂解液[22],含7 mol/L尿素、2 mol/L硫脲、4%(w/V)CHAPS、2% (V/V) Pharmalyte (pH 3~10)、1% (w/V) DTT(現用現加)。配制完成后分裝到Eppendorf管中,-20℃冰箱保存。
1.1.2儀器 雙光束紫外可見分光光度計UV-1800(日本島津)。
1.2 方法
1.2.1Bradford法線性范圍的確定 用BSA標準液分別配制含0~1 000 μg和0~200 μg標準蛋白質的樣品, 0~1 000 μg范圍內各樣品濃度依次為0.5 mg/mL、1.0 mg/mL、1.5 mg/mL、2.0 mg/mL、3.0 mg/mL、4.0 mg/mL、5.0 mg/mL、6.0 mg/mL、7.0 mg/mL、8.0 mg/mL、9.0 mg/mL和10.0 mg/mL;0~200 μg范圍內各樣品濃度依次為0.2 mg/mL、0.4 mg/mL、0.6 mg/mL、0.8 mg/mL、1.0 mg/mL、1.5 mg/mL 和2.0 mg/mL。測定吸光度時以上各樣品均取100 μL,對照樣品均為100 μL水(以對照樣品和5 mL顯色液的混合液進行儀器調零,下同)。按Bradford法[12]測量各樣品在595 nm處對應的吸光度值(A595)。測定時與樣品反應的顯色液體積均為5 mL,不同濃度樣品均設3個重復。
1.2.2不同體積lysis和不同lysis組分干擾程度的測定 首先,分別配制含10 μL、40 μL、70 μL、100 μL lysis的水溶液,各樣品體積均為100 μL,以100 μL水為對照樣品,測定A595,并計算相當于標準蛋白BSA的值(μg)。其次,分別用水配制7 mol/L 尿素、2 mol/L 硫脲、4%(w/V) CHAPS、2% (V/V) Pharmalyte (pH 3~10)、1% (w/V) DTT等含lysis單一成分的溶液,各取100 μL加入含5 mL顯色液的試管中,搖勻并反應5 min后測定A595,對照樣品為100 μL水。再次,分別配制4%的CHAPS、NP-40和Triton X-100溶液。測定100 μL上述溶液與5 mL顯色液反應后的A595,對照樣品為100 μL水。
1.2.3Lysis對蛋白質-染料復合物穩定性的影響
Bradford最初報道該方法時,測定了0~70 min內蛋白質-染料反應液吸光度的變化,并說明顯色反應在2 min時基本完成,A595在1 h內變化不會超過±4%[12],即變化幅度在8%左右。Kruger[20]在介紹Bradford法時也建議在混勻后2~60 min內測定A595。目前尚無其他文獻報道Bradford法的穩定性,也沒有lysis對蛋白質-染料復合物穩定性影響的報道。為研究lysis對蛋白質-染料復合物穩定性的影響,本研究分別測定了有無lysis條件下1 h內反應液A595的變化。
配制0.5 mg/mL的BSA標準液。測定無lysis條件下蛋白質-染料復合物的形成速率及穩定性時,取100 μL該標準液和100 μL水加入含有5 mL顯色液的試管中,搖勻后立即放入分光光度計中連續測定1 h內A595的變化,儀器每2 s自動記錄吸光度值,對照樣品為200 μL水。測定存在lysis條件下蛋白質-染料復合物的形成速率及穩定性時,取100 μL該標準液和100 μL lysis加入含有5 mL顯色液的試管中,搖勻后立即測定1 h內A595的變化,對照樣品為100 μL水和100 μL lysis的混合液。
1.2.4有無lysis兩種條件下0~50 μg內標準曲線的制作 蛋白質定量標準曲線的確定要考慮加樣體積和測定樣品中蛋白質的濃度范圍。經典Bradford方法[12]和實驗指導教材中[18]介紹的加樣體積均為100 μL。但2-DE樣品體積越大,由lysis造成的背景干擾越嚴重,因此可通過減少加樣體積來降低樣品中lysis的干擾,但加樣體積過小又會導致測量誤差的增加。
根據蛋白質干粉樣品在lysis中的溶解比例,2-DE樣品中蛋白質濃度一般在2~5 μg/μL之間[3,23]。若定量時加樣體積為100 μL,則樣品中蛋白質含量為200~500 μg。為避免定量時超出線性范圍,在測定時需要對樣品進行一系列的稀釋,操作較繁瑣,也浪費樣品。綜合考慮lysis干擾程度、測量誤差和操作便利性,將測定lysis溶解的蛋白質樣品的加樣體積優化調整為10 μL。加樣體積調整后,則10 μL樣品中約含有20~50 μg蛋白質,恰好在Bradford法的線性范圍內,無需稀釋,可直接測定。
分別用水配制含10 μg、20 μg、30 μg、40 μg、50 μg BSA的10 μL和100 μL標準蛋白樣品體系。測定10 μL lysis對標準曲線的影響時,向10 μL標準樣品體系中和對照樣品(10 μL水)各加入10 μL lysis;將各樣品加入5 mL顯色液中,反應完全后測定A595。測定100 μL lysis對標準曲線的影響時,向100 μL標準蛋白樣品體系和對照樣品(100 μL水)中各加入100 μL lysis,其他操作同上。
2 結果與分析
2.1 Bradford法的線性范圍
線性范圍是制作標準曲線的依據。現有文獻中報道的Bradford法的線性范圍是2~50 μg[19]或10~200 μg[18],差異較大,因此需要加以明確。如圖1A所示,在0~1 000 μg范圍內,A595隨著樣品中蛋白質含量的增加而增加,但當蛋白質含量超過900 μg后A595趨于平穩,說明Bradford法最大檢測量為1 mg左右,線性關系較好的區段在0~200 μg以內。通過在0~200 μg范圍內增加數據點,并進行0~100 μg、0~150 μg、0~200 μg范圍內數據的線性擬合,獲得各范圍內數據的線性相關系數R2分別為0.9991、0.9767、0.9567,表明Bradford法的線性范圍是0~100 μg,超過100 μg后線性關系逐漸變差。根據線性范圍內蛋白質含量與對應吸光度之間的關系,計算得出Bradford法的檢測限約為11 μg/mL。
2.2 Lysis及其成分對Bradford法的干擾
為探究lysis的背景干擾效應,對10~100 μL范圍內不同體積lysis與顯色液反應的A595進行了測定。如表1所示,lysis的干擾程度與其體積呈正相關(R2為0.986 2)。100 μL lysis與顯色液反應產生的背景相當于16.94 μg標準蛋白BSA與顯色液反應產生的吸光度。10 μL lysis造成的背景干擾僅為100 μL lysis的1/3。
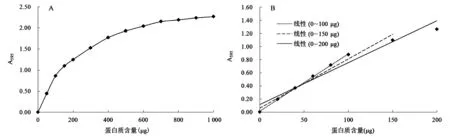
圖1 0~1 000 μg(A)及0~200 μg(B)范圍內蛋白質含量與A595的關系Fig.1 The relationship between protein content from 0 to 1 000 μg(A)and from 0 to 200 μg(B)and absorbance at 595 nm.
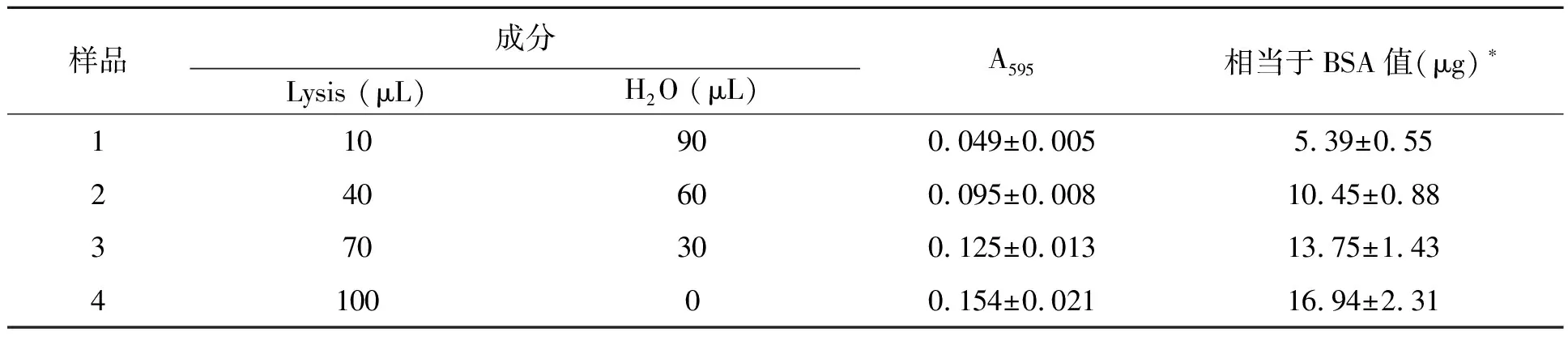
樣品成分Lysis (μL)H2O (μL)A595相當于BSA值(μg)?110900.049±0.0055.39±0.55240600.095±0.00810.45±0.88370300.125±0.01313.75±1.43410000.154±0.02116.94±2.31
*各樣品均與5 mL顯色液反應并測定595 nm處吸光度;根據0~100 μg范圍內標準曲線計算各吸光度對應的BSA值。
干擾Bradford法測定的化合物眾多,普遍認為表面活性劑是造成干擾的主要因素[12,24,25]。表面活性劑的作用是在不破壞蛋白質結構、保持生物活性的情況下改善蛋白質的溶解度。Lysis中的高濃度變性劑、表面活性劑、還原劑和兩性電解質等都可與顯色液發生反應,導致背景干擾(圖2)。結果顯示,兩性電解質Pharmalyte (pH 3~10)產生的背景干擾最大,其他組分產生的干擾較微弱,表明并不是所有表面活性劑都會嚴重干擾Bradford法。
Lysis中常用的表面活性劑有兩性離子型的CHAPS,以及非離子型的NP-40和Triton X-100等。圖3顯示不同表面活性劑之間差異極顯著。Triton X-100和NP-40引起的背景色極高,而CHAPS造成的背景干擾較小。100 μL 4% (w/V) Triton X-100和NP-40的A595(2.47)已經超出1 mg蛋白質的A595(Bradford法的最大檢測量,圖1),導致無法測出準確的蛋白質濃度。
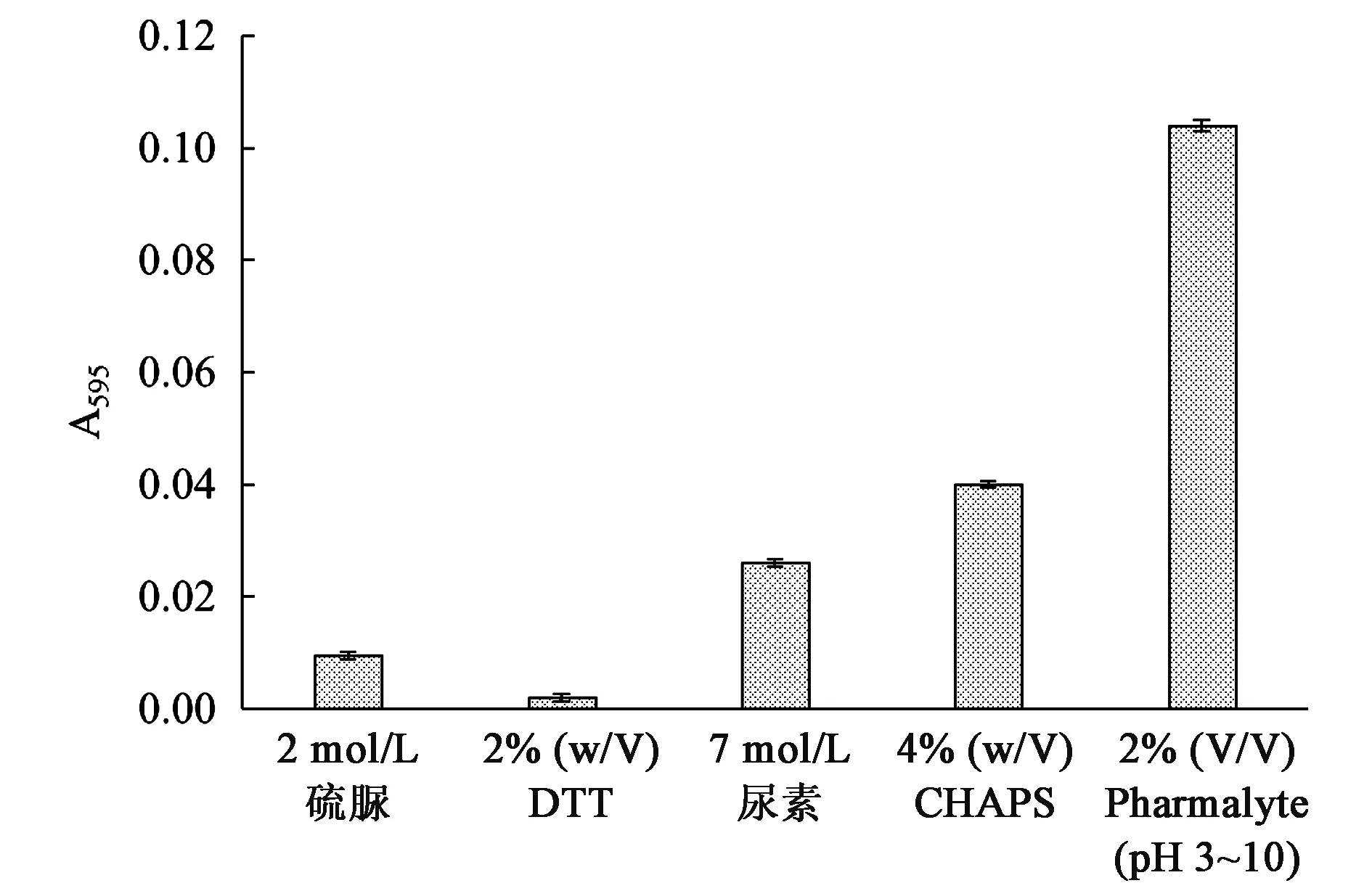
圖2 Lysis不同組分對Bradford法的干擾程度Fig.2 The interfering effect of different components of lysis on the Bradford assay.注:加樣體積均為100 μL。
2.3 Lysis對蛋白質-染料復合物穩定性的影響
為研究lysis對蛋白質-染料復合物穩定性的影響,本研究分別測定了有無lysis條件下1 h內反應液A595的變化。如圖4和表2所示,反應體系中無lysis時,A595從混勻后3 min開始基本趨于穩定,3~60 min內變化幅度為+2.82%到-0.54%(以180 s時A595為基準)。本研究采用的顯色液組分與經典Bradford法稍有不同,即磷酸含量從8.5%(w/V)提高到10.2%(w/V)。對比Bradford[12]的研究中0~70 min內蛋白質-染料穩定性圖譜可以發現,磷酸比例的增加極大提高了蛋白質-染料復合物的穩定性。
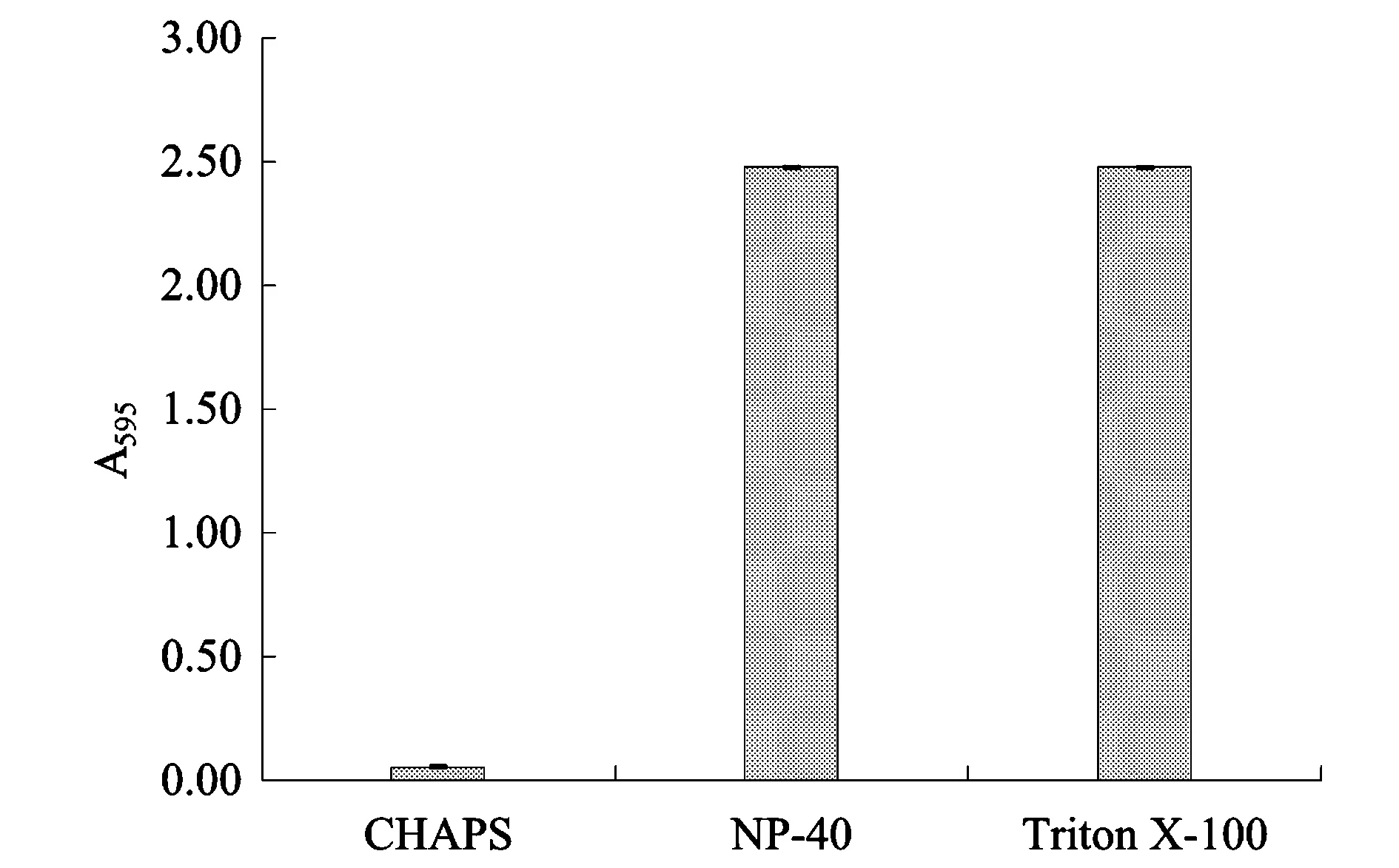
圖3 三種表面活性劑對Bradford法的干擾程度Fig.3 The interfering effect of three kinds of detergents on the Bradford assay.注:加樣體積均為100 μL,各表面活性劑濃度均為4%(w/V)。
若反應體系中存在lysis,則A595從混勻后2 min開始基本趨于穩定,之后緩慢上升至12 min左右,然后一直呈下降趨勢,2~60 min內變化幅度為+2.07%~-8.43%(以120 s時A595為基準),約是無lysis時變化幅度的3倍,從50 min開始A595出現劇烈波動。兩種條件下吸光度的變化表明lysis對蛋白質-染料復合物的穩定性有顯著影響。另外,比較圖4中兩條曲線可發現,對于相同蛋白質含量樣品,當反應液中存在lysis時測得的吸光度值顯著高于無lysis時,說明lysis會影響蛋白質和染料之間的顯色反應。
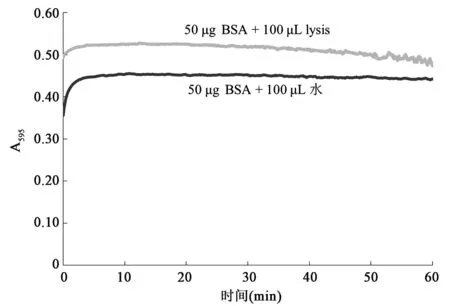
圖4 Lysis對蛋白質-染料復合物形成及穩定性的影響Fig.4 The effect of lysis on the formation and stability of protein-dye complex.
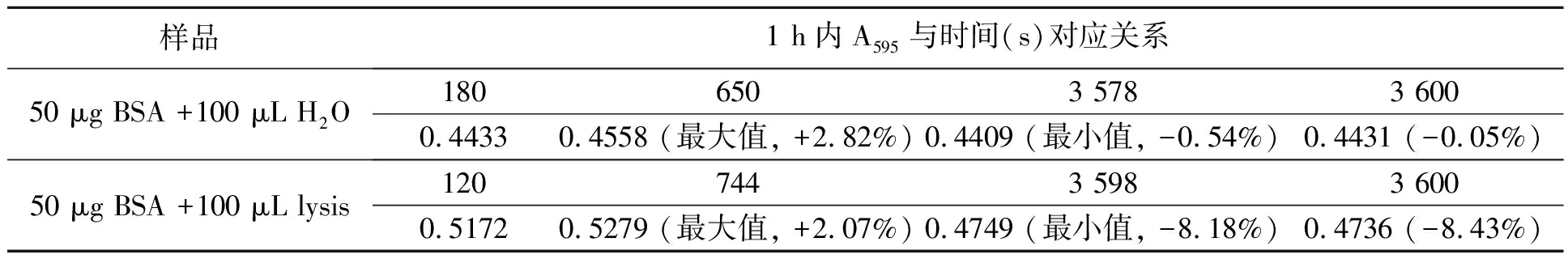
表2 有無lysis兩種條件下BSA與顯色液反應1 h內A595的變化Table2 Absorbance change at 595 nm of the mixture made by BSA and dye reagent,with or without lysis buffer in 1 h.
2.4 加樣體積調整及其對標準曲線的影響
圖5顯示了加樣體積調整對0~50 μg范圍內標準曲線的影響。當加樣體積為100 μL時,測量數據波動性較大,而且標準曲線的線性關系不佳(R2=0.996 8),可能與lysis的背景干擾較大有關。當加樣體積減少為10 μL時,測得數據穩定性更好,線性關系也有明顯改善(R2=0.999 1)。
3 討論
本研究通過分析蛋白質含量與吸光度的線性關系,確定Bradford法的線性范圍是0~100 μg,因此何忠效等[18]報道的線性范圍過大,而Okutucu[19]給出的線性范圍又偏小。當加樣體積為100 μL時,與線性范圍對應的濃度范圍是0~1 μg/μL;當加樣體積為10 μL時,對應的濃度范圍是0~10 μg/μL。在制作標準曲線時,應保證檢測范圍在線性范圍內。另外,由于標準蛋白質的純度不同,稱量和配制過程也會有誤差,為使不同研究人員的定量數據具有可比性,應對1 mg/mL的標準蛋白溶液進行校準。
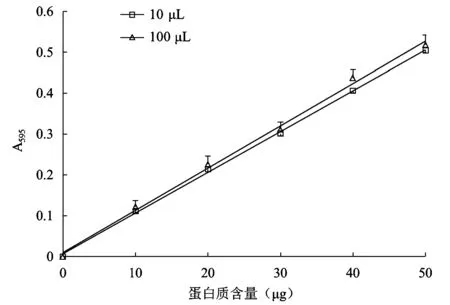
圖5 不同加樣體積所得0~50 μg蛋白質范圍內標準曲線Fig.5 Construction of the standard curve in the range of 0~50 μg(BSA)with different sample volume.
Bradford通過分析常用試劑對定量的影響,發現Bradford法對大多數常用化學物質的兼容性較好,但高濃度的表面活性劑(1%的Triton X-100和SDS)會嚴重干擾測定[12]。本研究發現并非所有表面活性劑都會產生嚴重干擾,CHAPS產生的背景要遠小于Triton X-100和NP-40。Lysis中表面活性劑的濃度高達4%(w/V),若應用Bradford法進行定量,在配制lysis時應避免使用Triton X-100和NP-40等,而選擇CHAPS或其他背景干擾較小的表面活性劑。
通過有無lysis條件下對蛋白質-染料復合物穩定性的分析可知,lysis會影響蛋白質-染料復合物的穩定性。對于溶解在水中的蛋白質,蛋白質-染料復合物在1 h內有很好的穩定性,但對于溶解在lysis中的蛋白質,蛋白質-染料復合物穩定性降低,在反應50 min以后尤其明顯。Bradford建議常規樣品在反應后2~60 min內測定吸光度[12];但定量含lysis的2-DE樣品時,適宜的測定時間為反應開始后3~30 min,該時間段內A595的變化幅度約為1%,對測量值的影響較小。由于提高顯色液中磷酸含量可顯著改善蛋白質-染料復合物的穩定性,因此定量2-DE樣品時可將磷酸含量從8.5%(w/V)提高至10.2%(w/V)。
由于lysis會影響蛋白質與染料的顯色反應,導致相同含量蛋白質在有無lysis條件下吸光度有較大差異,因此在測定2-DE樣品時不但要以相應體積lysis做對照來消除lysis的背景干擾,還要在制作標準曲線時考慮到lysis的影響,在標準蛋白樣品和對照中都加入相應體積的lysis,否則會使測定結果出現較大偏差。至于lysis影響蛋白質和染料相互作用的機制還需進一步研究。
經典Bradford法加樣體積為100 μL[12]。為使該法更適合定量2-DE樣品,將加樣體積調整為10 μL。減少加樣體積不但可以減少lysis導致的背景干擾,提高定量結果的準確性,還可以省去樣品稀釋步驟,簡化操作。對于難提取的膜蛋白和其他特殊蛋白樣品,也可節省樣品。
綜上所述,本研究通過分析Bradford法的線性范圍、測定lysis及其組分的干擾效應、分析lysis對蛋白質-染料復合物穩定性的影響和調整加樣體積,針對定量蛋白質組學2-DE樣品存在的主要問題對Bradford法進行了優化,可為蛋白質組學相關領域的研究人員快速掌握和應用該法提供參考。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
