EBV與TLRs信號通路相互作用機制研究進展
2019-05-28 03:46:40張旭娟趙鵬翔YAOMawulikplimiAdzavon李秦劍
生物技術進展 2019年3期
張旭娟, 趙鵬翔*, YAO Mawulikplimi Adzavon, 李秦劍, 謝 飛
1.北京工業大學生命科學與生物工程學院, 北京 100124;2.陸軍軍醫大學新橋醫院, 全軍血液病中心, 重慶 400037
EBV為人類皰疹病毒4型(human herpesvirus 4,HHV-4),屬于γ皰疹病毒亞科,于1964年由Epstein與Barr研究非洲兒童伯基特(Burkitt)淋巴瘤時發現[1]。成熟的EBV病毒顆粒呈球形,直徑約為180 nm,基因組為雙鏈線性DNA,長度約為172 kb,編碼基因約85個[2]。EBV在全球范圍內的人群感染率超過了90%[3],且感染人群主要為青少年。大多數原發性感染起初并無癥狀,但若于青春期或成年期感染,則更容易引發傳染性單核細胞增多癥[4]。原發性感染后,EBV會在B細胞中建立潛伏感染,并通過一系列的調控來逃避宿主TLRs等免疫系統的識別與免疫作用,在特定條件下,EBV被激活進入裂解感染期,產生子代病毒顆粒從而引發疾病。此外,EBV作為第一個發現的與腫瘤相關的病毒,與眾多腫瘤的發生有關,如Burkitt淋巴瘤、鼻咽癌、胃癌、宮頸癌和乳腺癌等[5]。然而,EBV也是溶瘤病毒。促癌和溶瘤這2種截然不同的作用主要是由EBV不同的侵染狀態所決定的,而TLRs信號通路能夠影響EBV侵染狀態從而影響EBV的生物學作用。
TLRs作為固有免疫中模式識別受體之一,一方面能夠識別EBV并控制病毒的感染,但TLRs的異常激活也會促進EBV的感染效率及其部分蛋白的表達;另一方面,不同感染狀態的EBV對TLRs表達水平的調控作用也各不相同。本課題組在眼眶炎性假瘤(idiopathic orbital inflammatory pseudotumor,IOIP)的臨床樣品以及EBV+的B淋巴瘤細胞系中發現EBV與TLRs的活化存在潛在聯系[6,7],提示深入研究TLRs與EBV的相互作用機理,將對臨床治療及實驗研究有所助益。基于此,本文對EBV與TLRs信號通路之間相互作用的研究進展進行了綜述,以期為開發新的EBV治療靶點以及完善TLRs激動劑的臨床方案提供一定的參考。
1 EBV生活周期
EBV的原發性感染一般發生在口咽的唾液交換過程,宿主為B淋巴細胞和特定上皮細胞等。EBV通過糖蛋白gp350和B細胞表面受體CD21促進EBV囊膜與細胞膜融合,使EBV外層基質及核衣殼進入細胞內。核衣殼轉運到核膜后,EBV DNA進入細胞核內,激活γ干擾素誘導蛋白16(γ interferon inducible protein 16,IFI16)下游的炎性小體通路以及DNA損傷反應、ATM激酶信號通路。而EBV線性雙鏈在其末端重復序列TR的介導下進行環化,進而被宿主組蛋白識別修飾,建立穩定的潛伏感染狀態,外界刺激或機體免疫力低下等條件會誘導EBV由潛伏期向裂解期轉化[8]。
1.1 潛伏感染
原發感染后的潛伏感染時期發生在記憶B細胞中,EBV通過一系列復雜的機制逃避宿主的免疫并在體內長期存在。該時期,EBV基因組通常是高度甲基化的,以環狀染色質結構穩定存在于宿主細胞核內,能夠利用宿主的DNA聚合酶隨宿主細胞周期進行復制,并在細胞質基質中可檢測到游離的雙鏈環狀病毒DNA[9]。潛伏感染主要分為4個階段,共表達近100個EB病毒基因和8種蛋白質,這些潛伏基因包括6個EB病毒核衣殼抗原(EBV nuclear antigens,EBNAs)基因、2個潛伏膜蛋白(latent membrane proteins,LMPs)基因、EBV編碼的2種小的非聚腺苷酰化RNA(EBV encoded RNAs,EBERs)和限制性內切酶BamHⅠ-A向右讀碼區的轉錄產物(BamHⅠ rightwards transcripts,BARTs),其中僅EBNA-1和LMP-1在裂解感染期也表達。潛伏期相關基因相應表達階段及其功能如表1所示,其中,LMP-1在潛伏期與裂解期均有表達,且作為病毒癌基因,越來越多的研究表明其與細胞的運動、轉移和腫瘤的侵襲相關。與EBV相關的惡性腫瘤主要由被潛伏感染的細胞組成,并有EBV編碼的轉化蛋白和非編碼的RNA表達。
1.2 裂解感染
在某些情況下,如TGF-β的誘導、缺氧環境、DNA損傷或宿主記憶B細胞分化為漿細胞的過程中發生的B細胞受體交聯等,EBV可能會重新活化進入裂解復制階段產生大量感染性病毒顆粒,并經口咽唾液在個體間傳播[28]。EBV在該階段的基因表達具有時序性,依次表達即刻早期基因(immediate-early genes,IE)、早期基因(early genes,E)和晚期基因(late genes,L)。首先轉錄的是即刻早期基因BZLF1和BRLF1,分別編碼轉錄因子Z(aka Z,ZTA,ZEBRA)和R蛋白(aka R,RTA),Z和R與EBV DNA裂解復制起始點結合,反式激活啟動子Zp(Z promoter)和Rp(R promoter),觸發早期基因的表達,編碼病毒DNA復制所需的蛋白質;隨著病毒基因組的復制,病毒晚期基因表達,并編碼病毒基因組包裝成感染性病毒顆粒所需的結構蛋白,即衣殼蛋白、基質蛋白、囊膜蛋白等。新包裝成的感染性病毒顆粒可通過胞吐或者在細胞裂解后釋放到細胞外。
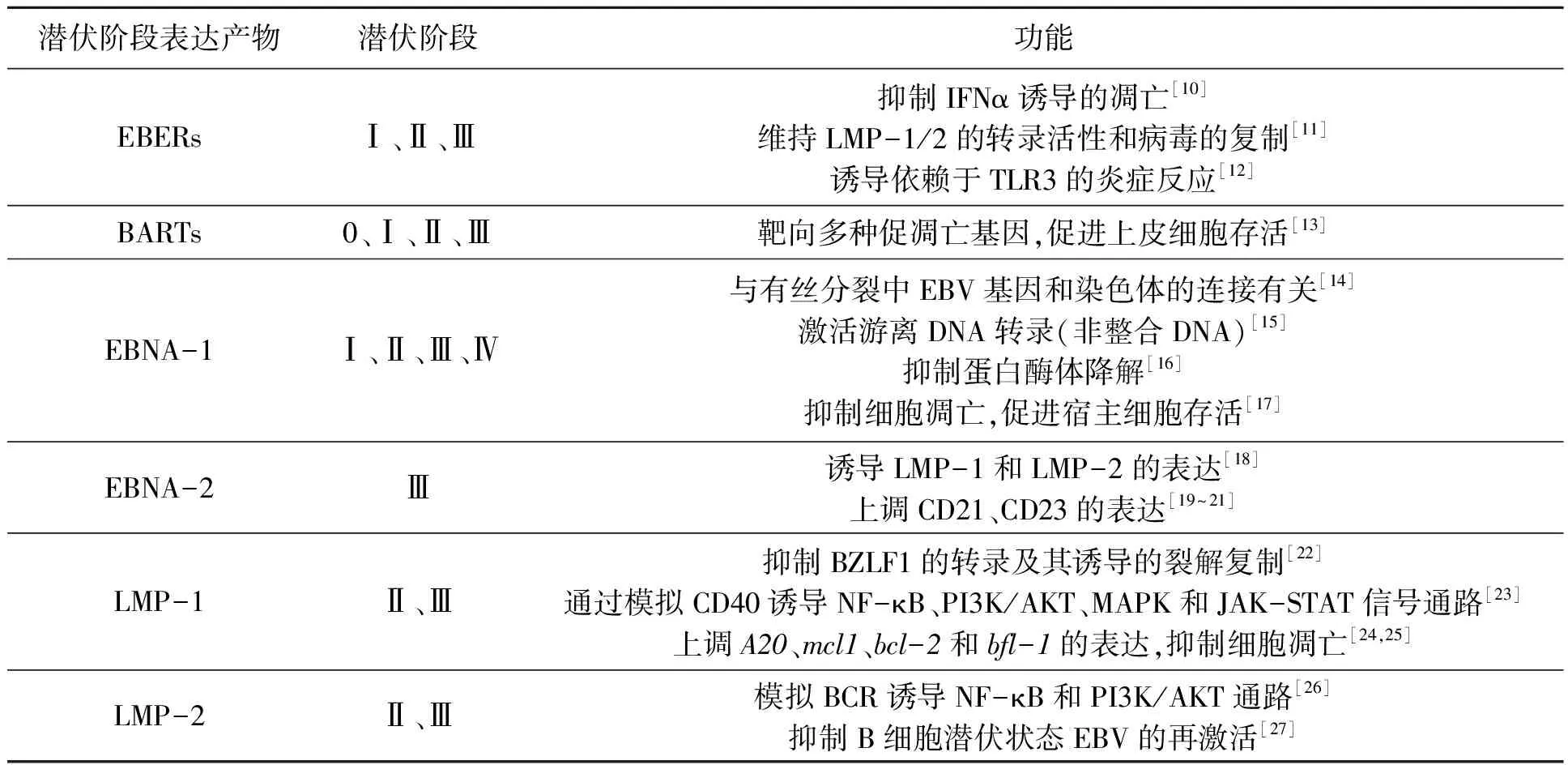
表1 EBV潛伏感染期相關部分基因及其產物Table1 Some related genes and their products of EBV at latent infection stage.

圖1 已知能夠調控Zp和Rp轉錄的因子Fig.1 Factors known to play roles in regulating transcription of Zp and Rp.注:順式作用元件用灰色矩形表示,其對應的反作用因子直接顯示在其上方,下方數字表示距離轉錄起始位點的堿基數目,其中轉錄起始位點為+1;YY1:轉錄因子Yin Yang-1;SP1/3:特異性蛋白1/3;SMADS:SMAD蛋白家族;MEF2D:肌細胞增強因子2D;CREB:環磷酸腺苷反應元件結合蛋白;ATFs:激活應激相關的轉錄因子;C/EBP:CCAAT增強子結合蛋白;c-Jun:翼式轉錄因子;ZEB1/2:鋅指結構轉錄因子1/2;EGR-1:早期生長反應因子1。
在整個EBV裂解周期中,BZLF1和BRLF1之間存在著復雜又緊密的聯系,它們的啟動子序列中存在很多宿主轉錄因子調控區域,受到嚴格的轉錄水平調控,目前已知的轉錄因子有:YY1、E2-2、JDP2、MEF-2D以及ZEB1/2等抑制Zp的轉錄;Sp1/Sp3、c-JUN/c-FOS、CREB/ATF/AP-1、SMAD以及C/EBP等激活Zp的轉錄[29];YY1和ZEBs抑制Rp的轉錄;EBR1和Sp1/Sp3激活Rp的轉錄。上述轉錄因子及其作用元件如圖1所示。
2 TLRs
在機體抵抗病毒的過程中,先天性免疫系統發揮著重要作用,可通過多種模式識別受體系統識別病毒,而TLRs作為先天性免疫系統的重要組成成分,是首個被定義的跨膜模式識別受體系統[30]。其存在于多種進化的生物中,能夠識別由微生物和內源性組織產生的信號,即通過病原體相關分子模式(pathogen-associated molecular patterns,PAMPs)和損傷相關分子模式(damage-associated molecular patterns,DAMPs),在信號轉導通路中發揮關鍵作用。不同類型的TLRs在識別先天性免疫病原體方面具有不同的功能,統稱為TLR家族,在哺乳動物中發現11種TLRs,其中,在人類中存在10種。先天免疫效應細胞可表達這10種TLRs,每種都與特定的配體結合,其中,TLR2識別酰化脂蛋白,TLR3識別雙鏈RNA,TLR4是脂多糖(lipopolysaccharides,LPS)的受體,TLR5識別細菌鞭毛蛋白,TLR7/8、TLR13識別單鏈RNA,TLR9識別雙鏈DNA,TLR10、TLR11識別寄生蟲抑制蛋白樣分子[31]。
2.1 TLRs相關信號通路
TLRs介導的信號通路分為髓樣分化因子88(myeloid differentiation factor 88,MyD88)依賴的信號通路和β-干擾素TIR結構域銜接蛋白(TIR-domain-containing adaptor inducing interferon-β,TRIF)依賴(或稱為MyD88非依賴)的信號通路,如TLR7/8/9屬于前者,TLR3屬于后者。MyD88依賴的信號通路中銜接蛋白MyD88招募下游的白細胞介素-1受體相關激酶(interleukin-1 receptor-associated kinases,IRAKs),將信號傳遞至腫瘤壞死因子受體相關因子6(TNF receptor associated factor 6,TRAF6),隨后募集并激活下游的轉化生長因子激酶1(transforming growth factor activated kinase-1,TAK1)和NF-κB抑制蛋白激酶α/β/γ(inhibitor of nuclear factor kappa-B kinase α/β/γ,IKKα/β/γ),促進AP-1活化以及NF-κB介導的炎癥因子(如TNF-α、IL-6、IL-8、IL-1α和IL-1β等)的表達[32,33]。TRIF依賴的信號通路中銜接蛋白TRIF招募下游的TRAF3,進而激活TBK1和IKKε,促進干擾素調節因子3/7(interferon regulatory factor 3/7,IRF3/7)所介導的Ⅰ型干擾素的產生[34]。而Ⅰ型干擾素能夠使JAK-STAT等信號通路活化,表達大量抗病毒基因,從而抑制病毒的復制[35]。上述TLR信號通路如圖2所示。
2.2 TLRs激動劑
TLRs激活介導產生的炎癥因子對機體有保護作用,其中大量研究證明其介導的腫瘤壞死因子具有抗癌作用,干擾素具有抗病毒作用;另有研究發現,人類免疫相關疾病中的TLRs表達異常升高,如系統性紅斑狼瘡患者的pDC細胞中TLR7的表達水平較高[37]。因此,TLRs激動劑除了可用于治療各種病原微生物引起的疾病,還可能成為自身免疫相關疾病的治療藥物。現階段,TLRs激動劑,如TLR3激動劑poly(I∶C)[38],TLR7激動劑R-848[39]、GS-9620[40]、852A[41]等,TLR9的激動劑CpG[42]等,大多數可作為免疫佐劑應用于臨床。TLRs除了能夠作用于免疫系統,也可作用于中樞神經系統,如激活中樞小膠質細胞的TLRs釋放IL-6等細胞因子,從而促進腦缺血的神經元存活,還能夠減少腦梗塞緩解腦血管疾病對大腦的損害[43];TLRs的激活狀態可影響小膠質細胞的吞噬能力,因此有望成為阿爾茨海默病免疫治療的潛在治療靶點[44]。
3 TLRs信號通路與EBV相互作用
TLRs作為模式識別受體中非常重要的一部分,可識別EBV并引發固有免疫,進而在一定程度上控制病毒的感染并促進在EBV潛伏感染狀態下B細胞的增殖;另一方面,越來越多的研究證明,EBV可能通過干擾TLRs的表達和功能,從而影響宿主免疫反應,造成免疫逃逸與持續潛伏感染,但目前對于TLRs與EBV之間的具體相互作用機制仍不完全明確。
3.1 EBV對TLRs信號通路的調控
EBV可以通過一系列的途徑降低TLRs的表達、逃避宿主固有免疫,從而建立并維持體內潛伏感染。人B細胞可表達高水平的TLR1、TLR6、TLR7、TLR9、TLR10,其中TLR7和TLR9的表達和功能與B細胞中EBV的關系較為密切。已有數據表明,EBV與TLR7、TLR9之間存在致病性串擾,EBV能夠影響或依賴TLR7、TLR9的表達,促進宿主B細胞的存活[45];與此同時,EBV通過抑制TLRs下游抗病毒因子的表達及功能而逃避宿主的免疫殺傷作用,由此可能引發B細胞功能障礙和免疫功能異常,從而引起一系列相關疾病。
EBV在不同的細胞中顯示出對TLRs具有不同的影響。研究表明,EBV能夠在體外感染纖維細胞并增加TLR7/9的mRNA表達水平[46];EBV也能夠誘導外周單個核細胞(peripheral blood mononuclear cell,PBMC)中TLR7/8的表達,但卻抑制TLR7/8/9對PBMC的增殖作用,這一增殖抑制作用對于外周血富集B細胞更為明顯,且呈EBV劑量依賴性[47,48];與此相悖,在幼稚B細胞中,EBV上調TLR7并下調TLR9的表達,使用TLR7激動劑R837與EBV共培養后,結果發現幼稚B細胞的增殖水平增加17倍[48],說明在幼稚B細胞中EBV并不能像在PBMC及外周血富集B細胞中一樣發揮抑制TLR7及其促細胞增殖的能力。Wang等[49]在研究自免型疾病系統性紅斑狼瘡(systemic lupus erythematosus,SLE)時發現,LMP2A可通過增強對TLR配體的反應活性,促進BCR/TLR通路對B細胞的自反應激活。由此可見,在不同細胞中EBV對不同的TLRs組分表達水平的調控作用并不完全一樣。此外,EBV對TLRs的細胞增殖作用的影響也不盡相同,這可能是由于不同細胞中基因選擇性表達形成不同的免疫應答機制,從而使EBV對TLRs的影響效應也存在差異。
EBV除影響TLRs對細胞的增殖能力外,還可通過影響TLRs下游信號通路(如Ⅰ型干擾素、IRF-5/7以及NF-κB等)來干預宿主的免疫功能或抗病毒能力。EBV感染漿細胞樣樹突狀細胞(plasmacytoid dendritic cells,pDCs)后能夠激活不成熟的pDCs,釋放受損性TNF-α,從而使機體無法啟動完整的T細胞免疫[50];EBV通過刺激pDCs誘導TLRs產生Ⅰ型干擾素,從而促進自然殺傷細胞和CD3 T細胞的活化,而此作用部分是由TLR9介導的[51];EBV感染的B細胞中釋放的EBER,主要負責EBV免疫激活,誘導TLRs下游Ⅰ型IFN和促炎細胞因子的釋放,從而可能引發免疫病理性疾病[52];Martin等[53]發現EBV能夠限制TLR7下游IRF-5對EBV建立持久感染的負作用;Dong等[54]的研究表明,在大多數EBV轉化的細胞中,IRF-5啟動子A區域存在高度甲基化,EBV編碼的蛋白BGLF4[55]以及BPLF1[56]都會抑制B細胞內TLRs介導的NF-κB活化,從而使EBV通過抑制TLRs下游信號通路抗病毒的能力而逃脫宿主固有免疫;不僅如此,EBV還可介導T細胞抑制因子B7-H1和ICOS-L的表達上調[51]。以上研究均說明EBV能夠通過TLRs及其下游信號通路間接或直接影響宿主或機體對EBV的免疫作用,進而維持自身的持續感染狀態。
在潛伏、裂解等不同感染時期,EBV基因及其產物的表達水平和功能各不相同,在EBV不同感染時期的B細胞中,TLRs的表達水平也具有差異性。在潛伏感染期中,隨著潛伏基因的表達,TLR6、TLR7、TLR9表達不變,TLR1和TLR10表達下降;但在被裂解感染的B細胞中,TLR1、TLR6、TLR7、TLR9、TLR10的mRNA表達水平均下降,尤其TLR9下降最為顯著[57]。造成TLR9被抑制的原因,目前主要集中在以下3個方面:①EBV表達的LMP1可通過激活NF-κB下調TLR9的轉錄[58];②B細胞中EBV重新激活后,裂解期蛋白BGLF5隨著病毒的再次活化下調TLR9的mRNA以及蛋白的表達[57];③然而也有研究者指出,EBV裂解感染后期表達的蛋白BPLF1能夠發揮去泛素化酶的作用,從而抑制TLRs介導的NF-κB在TRAF6信號通路中的激活[56]。除B細胞外,EBV在體外感染硬皮病SSc纖維細胞時會上調TLR7/9及IRF5/7的mRNA表達水平,但在細胞內無EBV時使用TLR激動劑并沒有引發TGF-β響應,由此推測,EBV在硬皮病SSc纖維細胞中可能作為一種弱誘導刺激產生了不完全的IFN信號[47]。可見,EBV潛伏期和裂解期對宿主固有免疫TLRs的調控作用并不一致。在EBV的活化過程中,對TLR9的影響格外顯著,這可能是由于在宿主抵抗病毒時,TLR9識別進入內涵體中的病毒雙鏈DNA,從而能夠在病毒復制過程中發揮出更強大的抗病毒作用。
最新研究還發現,EBV病毒蛋白可通過TLRs介導的信號誘導人視網膜色素上皮細胞使IL-6、IL-8、MCP-1分泌增加而引發炎癥,通過誘導NO合酶,合成NO以誘導視網膜細胞壞死和死亡[59],這一發現為了解EBV誘導炎癥和細胞死亡的免疫機制提供了重要信息,并有助于設計新的免疫治療方法來治療由EBV引起的眼部感染。
在長期的進化中,EBV與宿主固有免疫系統的相互作用可能使EBV已經發展出一系列類似挾持宿主以逃脫宿主的固有免疫的機制,但仍有待于進一步探究與闡明。
3.2 TLRs信號通路對EBV的不同效應機制
TLRs除了參與固有免疫過程限制EBV裂解感染,其他具體作用機制尚不明確,不同的TLRs可能發揮了不同的作用。TLR7/8激動劑R848、TLR9的激動劑CpG在CD40L的共同刺激下會顯著增加EBV對PBMC或者富集后B細胞的感染效率[60]。TLR7的異常激活可增加EBV LMP1的表達,TLR3、TLR 9激活產生的IRF7也參與了此過程[61]。TLR9的激活會抑制EBV裂解期基因BZLF1及其蛋白ZEBRA的表達,且這種抑制作用是不需要蛋白質從頭合成介導的,但需要功能性IRAK4、TLR9和MyD88蛋白的參與[62];TLR9觸發的BZLF1的組蛋白修飾也是抑制EBV裂解感染的重要因素,但NF-κB或其他已知的TLR9通路可能都不是TLR9介導抑制EBV的關鍵[63]。
TLRs下游信號通路中干擾素調節因子IRF-7與EBV介導的B淋巴細胞體外及體內轉化密切相關。近期研究發現IRF-7的下調會導致EBV轉化細胞生長受抑制,敲除IRF-7后細胞增殖水平降低,凋亡率上升,而當外源性表達IRF-7的質粒轉入后即可恢復病毒轉化細胞的生長;進一步研究發現IRF-7的下調會導致EBV的LMP1表達水平降低[64]。這表明IRF-7可能是EBV感染細胞轉化的一個因素,并可能成為治療EBV介導的瘤變的潛在靶點。
此外,還有研究證實TLR7/9在小鼠內皮細胞中參與識別了EBV DNA,但也因此觸發IL-17A的釋放引發炎癥[65]。由此可見,TLRs在識別控制EBV感染的同時對自身可能會造成如自身性免疫疾病等負面影響。這對評估TLR7/9在EBV陽性的自身免疫疾病的患者中的作用具有一定的指導意義。
雖然,TLR9觸發能夠抑制B淋巴瘤細胞中EBV向裂解感染的轉化,但也有研究發現在各種B淋巴瘤細胞中EBV的狀態并不決定細胞對TLR9所觸發的反應。使用TLR9的配體CpG2006激活TLR9后,能夠引發EBV宿主B淋巴瘤細胞的死亡,這一過程的發生依賴于半胱天冬酶,但與EBV及細胞周期均無關[66]。這一結果提示,B淋巴瘤細胞中TLR9的激活雖然能控制EBV的裂解性感染,但也可能會對宿主細胞產生負面傷害。
目前,TLRs多態性與EBV易感性的相關性研究,主要集中在TLR1、TLR2、TLR4、TLR7、TLR9的多態性位點,但實驗結果證明已報道的TLRs的多態性位點,如TLR1的rs5743551、rs5743611,TLR2的rs3804099、rs3804100[67],TLR4的rs1927911等,TLR6的rs3775073等,TLR7的rs2897827,TLR8的rs3764879、rs3764880,TLR9的rs187084、rs352140[68]等,與EBV易感性并無顯著相關性[69~72]。由此可見,TLRs的基因多態性并不是造成EBV易感與否的主要原因,相關機理探究可考慮嘗試TLRs多態性之外的其他因素。
本課題組對于眼眶炎性假瘤(idiopathic orbital inflammatory pseudotumor,IOIP)研究時發現,表達譜芯片測序與多組學分析結果均顯示TLRs通路呈活化狀態(數據未發表),同時在臨床樣品中檢測到了EBV-DNA和EBV相關抗原,提示IOIP的發病機理可能與EBV感染有關[6],且IOIP患者可能成為EBV誘發相關疾病的高危人群,需密切關注其EBV的感染狀態。
4 展望
作為全球范圍內高感染率的病毒,對EBV的研究越來越深入,研究方向大致分為以下4個方面:①EBV各成分如基因、蛋白以及一些小RNA的功能研究;②EBV與宿主之間的相互調控關系;③EBV的免疫逃逸機制;④EBV在相關疾病的發生及發展中的作用。
目前,有關EBV大部分研究集中于對其生活周期的調控,但宿主固有免疫系統與EBV之間的相互作用關系尚不完全明確,尤其是TLRs信號通路與EBV之間的相互影響遠比目前所了解到的更為復雜。TLR7/8/9具有交叉網絡的下游通路,盡管大部分研究著重探索了TLR9(識別DNA),但TLR7/8很有可能與EBV也存在潛在的相互作用。部分B細胞抗原受體(B cell antigen receptor,BCR)激動劑已經證實能夠激活潛伏狀態的EBV病毒,而TLR7/8/9的配體作為常用的免疫激動劑和抗體佐劑,在激活天然免疫的同時,是否會對大量人群中所攜帶的潛伏狀態的EBV的生活周期產生影響,可能是值得探討的重要問題。目前,HIV治療中已經應用shock-and-kill的新方法,有望徹底治愈HIV感染,該方法應用免疫激動劑,將T細胞中潛伏的HIV激活出來,從而再通過免疫系統將其清除。以這種方法為線索,對于正常人群中已經處于高概率潛伏狀態的EBV和巨細胞病毒等,通過外源激活的方法,徹底清除潛伏病毒,是將來可能的治療策略。而EBV等潛伏病毒,具有整合在宿主DNA,并且持續表達多種潛伏蛋白的特點,這些病毒組分通常也是造成細胞永生化和腫瘤生成的重要因素,因此,去除潛伏的EBV感染可能是未來治愈諸如伯基特淋巴瘤等惡性腫瘤的可能手段。EBV各時期基因、蛋白與TLRs相關通路的作用關系及關鍵分子的研究僅處于初步階段;此外,宿主對EBV周期調控基因轉錄翻譯的影響、EBV在宿主內維持感染以及在宿主間的轉移的具體機制,均有待于進一步研究。深入了解TLRs與EBV之間的限制關系有助于進一步理解EBV的生活周期,對于闡明EBV相關疾病發病機理以及篩選針對EBV的新型抗病毒疫苗和治療手段也具有重要意義,并且相關研究可能提示EBV參與的TLRs相關性自身性免疫疾病的新型治療靶點。
綜上所述,目前有關EBV與宿主之間相互作用的途徑和調控機制及其致病機制的相關研究并不詳盡,從而限制了抗EBV藥物的研發及臨床治療EBV相關疾病的推進。因此,深入理解EBV與宿主細胞固有免疫TLRs信號通路之間錯綜復雜的關系,對于闡明EBV相關疾病的發病機理和EBV相關疾病的診斷、新型治療方案的制定,具有重要的臨床應用價值。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
汽車工程學報(2017年2期)2017-07-05 08:13:02
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
