以帕金森癥狀起病的脊髓小腦共濟失調17型1例報告
2019-05-08 01:16:50梁慧胡詩俊劉濤文國強文剛
中國神經精神疾病雜志 2019年3期
關鍵詞:癥狀
梁慧 胡詩俊 劉濤 文國強文剛
* 海南省人民醫院神經內科(海口 570311)
△ 南方醫科大學南方醫院急診科
脊髓小腦共濟失調 17型(spinocerebellar ataxia 17)是一種少見的常染色體顯性遺傳性小腦共濟失調,臨床表現類似于亨廷頓病、肌張力障礙與帕金森病等多種疾病[1]。SCA17是由6號染色體上的TATA-box結合蛋白(TBP)基因編碼區的CAG/CAA重復數擴增所致。正常的等位基因為25~40次CAG/CAA,50次或以上CAG/CAA重復數為完全外顯等位基因,介于43~49次CAG/CAA的等位基因表現為部分外顯(約50%~80%)。到目前為止還不清楚41~42次CAG/CAA重復數是不是導致本病的低外顯率等位基因,但是近些年陸續有少許關于41~42次重復數所致該病的相關報道[2,3]。我們報道1例攜帶42次CAG/CAA重復擴增數的SCA17患者,臨床表現包括帕金森癥狀、進行性加重的共濟失調、自主神經功能損害,類似于多系統萎縮(multiple system atrophy,MSA)-P型起病并逐漸累及多個系統的MSA。
1 資料
患者,女,44歲,因“肢體活動不靈活 2年”于2017年11月3日入住海南省人民醫院神經內科。患者于2年前無明顯誘因開始出現左側肢體活動不靈活及肢體乏力感,伴有左側肢體抖動,靜止與活動時均出現,同時伴有運動遲緩,外院診斷為“帕金森病”,予“普拉克索及多巴絲肼片”控制癥狀。幾天后因頭暈停用普拉克索,繼續服用多巴絲肼1個月余,服藥期間癥狀無緩解,后因腎功能損害停用多巴絲肼。患者逐漸出現右側肢體活動不靈活、面容呆板、行走不便、講話費力等,未診治。5個月前因站立時頭暈及肢體活動不靈癥狀加重在外院診斷為“帕金森疊加綜合征”,未予特殊治療,癥狀逐漸加重,為求進一步診治收入我科。患者既往有“腰椎間盤突出”病史;否認高血壓、糖尿病等病史;年輕時從事美容行業,近10年當老板已基本不接觸美容產品;否認接觸毒物、抽煙、喝酒史等;否認服藥史。家族史無特殊。檢查:生命體征平穩,心肺腹未見明顯異常。神清,偶見患者突然強笑或強哭,均持續約1~2 min,構音不清,聲音低沉,對答切題,高級智能查體無異常。面具臉,雙側眼球活動正常,可見有輕度水平及垂直眼震,雙側瞳孔等大等圓,對光反射靈敏。口角無歪斜,伸舌居中,舌肌震顫,雙側軟腭上抬可,懸雍垂右偏,咽反射減弱。頸軟,左側肢體肌力4級,右側肢體肌力5級,四肢肌張力呈齒輪樣增高,以左側肢體為主,四肢腱反射活躍,雙側掌頜反射陽性,左側Babinski's征陰性,右側Babinski's征自發陽性,感覺系統檢查無異常,雙側輪替試驗、指鼻試驗、跟膝脛試驗均不穩,閉目難立征睜閉眼均陽性。
輔助檢查:臥位血壓116 mmHg/75 mmHg,立位(0 min)81 mmHg/48 mmHg,立位(3 min)79 mmHg/44 mmHg;血常規、肝腎功能、電解質、凝血功能、腫瘤標志物、免疫指標、甲亢七項、乳酸、維生素B12、輸血四項、血清銅及銅藍蛋白均未見明顯異常。尿常規:紅細胞計數 89個/μL↑、白細胞計數 1015個/μL↑、細菌 29043個/μL↑;同型半胱胺酸 20.9 μmol/L↑、葉酸 6.30 nmol/L↓。MMSE量表評分29/30分。腹部、頸部血管及心臟彩超未見明顯異常。甲狀腺彩超:甲狀腺右葉低回聲結節,考慮結節性甲狀腺腫。泌尿系彩超及殘余尿示右腎結石,膀胱內殘余尿量約18 mL。頭顱MRI可見全腦萎縮,小腦及腦干萎縮明顯,腦橋可見可疑十字征(圖1);頸椎MRI未見脊髓變細。
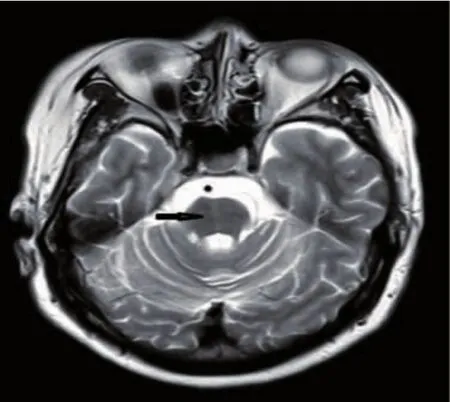
圖1 頭顱MRI腦干可見可疑十字征(黑箭頭所示)
基因檢測(廣州金域醫學檢驗中心):MSA的SNCA基因測序未見異常。SCA17相關基因CAG/CAA重復數:其中一個為36次,另一個為42次。其他亞型 SCA1、SCA2、SCA3、SCA6、SCA7、SCA8、SCA10、SCA12、齒狀核紅核蒼白球路易體萎縮相應基因及Friedreich’s共濟失調基因重復片段的重復數目均在正常范圍。
2 討論
SCA17臨床診斷復雜,頭顱MRI大多數表現為大腦、小腦和腦干不同程度的萎縮。典型的共濟失調和構音障礙是最突出的臨床表現,其他表現包括人格改變、精神異常、認知障礙和基底節功能障礙等[4]。因此,SCA17也被定義為類亨廷頓樣疾病。由于SCA17表型與其他神經變性疾病如亨廷頓病和其他類型SCA類似,僅憑臨床癥狀來診斷SCA17是十分困難的。SCA17的發病年齡大約在3~55歲之間,此病發病年齡在一定程度上與CAG重復次數有關,但不如其他的多聚谷氨酰胺疾病如亨廷頓病明顯[5]。發病年紀較晚及疾病進展程度較慢的溫和表型一般與CAG重復次數較短(42~44次)有關,而較長CAG重復次數的患者臨床表現更嚴重和進展更快。有研究認為SCA17低重復擴增數更容易引起帕金森樣癥狀而不是共濟失調。
到目前為止,SCA17病理性CAG重復擴增數尚未完全明確。許多報道認為43次或以上CAG重復擴增數為異常范圍,但近期越來越多的研究表明41次和42次CAG重復擴增數均可致病[6]。MD[7]等報道6例SCA17重復擴增數42次并以帕金森癥狀為主要表現的患者,起病年齡56~68歲,均無家族史,其中5例患者被診斷為帕金森病,1例患者被診斷為MSA;因此低重復擴增數SCA17患者(尤其是42次)可能大部分以帕金森樣癥狀起病,符合我們此例患者。LEE[2]等報道了1例CAG重復擴增數42次患者56歲開始出現構音障礙、走路不穩、眼球掃視及病理性強哭強笑。DOHERTY[8]等報道1例CAG擴增數41次患者59歲時以晚發型共濟失調起病。和此例患者一樣,上述所報道的SCA17患者均是經過了數年時間且排除其他獲得性或遺傳性運動障礙疾病后才診斷出來的。SCA17廣泛的臨床譜給此病的診斷帶來了一定的難度,在這些病例中雖患者攜帶的CAG重復次數一樣,但患者的起病年齡和癥狀、疾病的進展過程均不相同。另外,可能低次數的TBP重復序列表現為不完全外顯率,故大部分患者無相關追溯的家族史,進而容易導致此病的漏診。
自主神經功能障礙是MSA的特點之一,但在多種脊髓小腦共濟失調中包括SCA17中均有可能存在自主神經功能障礙[9];十字征常見于 MSA,但并非特異性表現,在SCA2、3、7、8和17中均可能出現。LEE等曾報道診斷為SCA17且CAG重復序列為42次的患者中出現與情緒無關且無法控制的病理性強哭強笑[2],此例患者亦出現病理性強哭強笑,之前的研究認為病理性強哭強笑是由于大腦-腦橋-小腦通路受損所致,與情緒執行能力相關的小腦結構受損可能導致異常的哭笑。因此,病理性強哭強笑可能是SCA17不典型的臨床表現,可能與小腦和小腦外萎縮有關。
此例患者定位診斷:患者肢體活動緩慢,四肢肌張力增高、面具臉及運動遲緩,定位在黑質紋狀體;患者站立不穩,指鼻試驗、跟膝脛試驗欠穩準,聲音低沉及眼球震顫定位在小腦;立位血壓較臥位血壓≥30 mmHg/15 mmHg及泌尿系彩超可見殘余尿,定位在自主神經功能;雙側掌頜反射陽性、四肢腱反射活躍,定位在雙側錐體束。定性診斷:患者中年女性,慢性起病,逐漸進展加重,考慮為變性、腫瘤、中毒或遺傳;頭顱MRI未見明顯占位,可排除腫瘤;患者既往曾從事美容行業10年,近10年已基本不接觸相關產品,余無毒物接觸史,考慮中毒可能性很小,故重點考慮變性或遺傳;根據患者起病形式、癥狀及頭顱MRI可見可疑十字征,最初我們診斷患者很可能是MSA,但患者存在強哭強笑及錐體束明顯受損體征,我們不排除患者為SCA,患者雖否認家族史,但父母親已過世無法證實是否為相關基因攜帶者,不排除小孩為致病基因攜帶,目前尚不發病,遺憾的是患者愛人不同意予孩子進一步基因檢測。行SCA相關基因檢測后發現TBP基因的CAG/CAA重復數為42次和36次,最終診斷為SCA17型。
當遇到臨床表現類似MSA患者時,SCA17應被列為鑒別診斷之一,特別是對于那些表現不典型的患者。CAG 42重復擴增數的患者疾病進展過程相對緩慢,但我們這例患者卻不然,該患者雖然CAG重復次數僅42次,但表現為典型的表型如帕金森癥狀和小腦共濟失調等。王俊玲[10]等研究表明我國漢族人群中正常SCA17最大的三核苷酸重復次數為41次,因此,對于SCA17致病基因多核苷的重復次數還需要進一步研究。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
