伴有腦白質病變的強直性肌營養不良
2019-05-08 01:16:48路延朋李端孫舒研宋學琴吳紅然張慧卿孫瑜
中國神經精神疾病雜志 2019年3期
路延朋 李端 孫舒研 宋學琴 吳紅然 張慧卿 孫瑜
強直性肌營養不良 (myotonic dystrophy,MD)是一種最常見的成人型肌營養不良,臨床上主要表現為進行性肌無力、肌萎縮、肌強直,除骨骼肌受累外,還累及其他多個系統,包括心臟,眼睛,內分泌系統,中樞和周圍神經系統,平滑肌,骨骼和皮膚等[1];其遺傳方式為常染色體顯性遺傳,臨床上主要分為兩型:MD1、MD2;MD1包括成人型,先天型,早發型,晚發型;成人型,即經典型,臨床上最常見[2]。關于DM1中樞神經系統病變的研究國內較少,本文就1例伴有核磁T2、FLAIR白質病變的DM1患者進行分析及文獻復習,以提高廣大同道對該病變的認識。
1 臨床資料
患者,女,33歲。因“頭痛、發熱4 d”入院。患者緣于4 d前因受涼后出現頭痛,以左側顳部為著,呈波動性,伴惡心、嘔吐,伴發熱,體溫最高達38.6℃,無寒戰;既往剖宮產術后3個月;查體:體溫 38.1℃ ,雙肺叩清,呼吸音清,未聞及干濕性啰音。神清,記憶力、計算力差,四肢肌力5級,雙側Babinski’s 征可疑陽性,雙劃征(+),余神經系統查體未見異常。輔助檢查;血常規、凝血常規、尿常規、便常規未見異常,生化全項:肌紅蛋白:103.0 ng/mL、肌酸激酶:928.0 U/L、乳酸脫氫酶:281.0 U/L、低密度脂蛋白膽固醇:3.45 mmol/L;行頭顱CT示:雙側基底節區及雙額葉小片低密度、腦干可疑小片少低密度。腦電圖示:廣泛中度異常;腰椎穿刺術:測腦脊液壓力為100 mmH2O,腦脊液生化:蛋白:0.40 g/L;腦脊液細胞學示:可見20個淋巴細胞、8個單核細胞、40個中性粒細胞、4個激活性單核細胞;自身免疫性腦炎抗體陰性;頭MR平掃+增強:雙側額頂顳葉、左側海馬區可見多發斑片狀、片狀等/稍長T1長T2高FLAIR信號,邊緣尚清,呈較對稱性分布;雙側側腦室周圍可見暈狀稍長T1長T2高FLAIR信號,建議遺傳學檢查,病灶未見強化 (圖1 B);SWI未見出血灶;蒙特利爾認知評估量表 (Montreal cognitive assessment,MoCA):包括畫鐘實驗、視空間與執行功能、命名、注意力、語言能力(重復、流暢性)、延遲回憶、定向力;其中視空間與執行功能重度失分(0/2)、畫鐘實驗失分(2/3)、注意力失分(2/6)、語言能力里的重復能力重度失分(0/2)、抽象評分重度失分(0/2)、延遲回憶(4/5)和定向力(5/6)失分,評分為:17分(滿分30分),患者為初中學歷,≤19分為異常;簡易智能精神狀態檢查量表(minimental state examination,MMSE): 包括定向力、記憶力、注意力/計算力、回憶能力、語言能力(命名能力、復述能力、三步命令、閱讀能力、書寫能力、結構能力);其中定向力失分(8/10)、計算力重度失分(0/5)、回憶能力失分(2/3)、語言能力中的三步命令和結構能力失分 (7/9),該患者評分為20分(總分30分),≤24分為異常;患者核磁顳極及外囊部位可見白質病變,伴有認知功能異常,伴有頭痛,疑似患者為CADASIL,后追問患者家族史,患者訴家族中10人均伴有反應較差,行走不穩,握拳后手指不能伸直癥狀,癥狀均較患者本人差;其中有3人伴有白內障病史;患者育有2子,1子自出生不哭,且活動差,走路跌跤,存在持久握拳后手指不能伸直,目前8歲,家屬訴自小智力差,目前仍未上學;家族中無卒中史,無偏頭痛病史,家族成員發病較早,不符合CADASIL;再次查體發現患者雙手大魚際肌叩擊后有肌球形成,查肌電圖+神經傳導速度:被檢肌可見大量肌強直電位;左腓總神經復合肌肉動作電位波幅減低;眼科檢查:雙眼白內障;肌肉活檢結果:HE染色可見肌束衣和肌內衣結締組織和脂肪組織部分區域輕度增生,肌束內肌纖維部分區域排列較疏松,肌纖維大小不一,可見多數萎縮和少數肥大纖維,明顯核內移和核袋(圖1A),偶見肌纖維變性、肌分裂和肌再生;考慮患者為強直性肌營養不良,行基因檢測:DMPK基因3′UTR區的CTG重復序列超過50次。綜上最終診斷該患者為:強直性肌營養不良1型,家族為肌營養不良家系。
2 討論
MD1由肌營養不良蛋白激酶基因(myotonic dystrophy protein kinase,DMPK)非翻譯區的三核苷酸CTG異常重復所致;該基因位于染色體19 q35,其常見的異常重復序列為50~4000次;MD2由鋅指蛋白9基因上的第一個內含子(zinc finger protein 9,ZNF9)中的核苷酸 CCTG異常重復所致。大量的數據表明DMPK的RNA包含著異常重復的CUG被保留在細胞核內,干擾了RNA的蛋白結合功能,從而導致了RNA的剪切異常。
DM1臨床上主要表現為肌無力、肌萎縮、肌強直,其他系統的表現包括糖尿病、心臟病、周圍神經病、禿頂、白內障等,本例患者經眼科檢查伴有雙眼白內障;白內障可為DM1患者早發的唯一癥狀[4];但臨床上的表現具有高度異質性,可從嚴重的,通常是致命的到無癥狀的遺傳缺陷攜帶者;有研究顯示,該病存在遺傳早現現象[3]。文獻中有報道強直性肌營養不良患者有類似中風樣發作[5],其與患者伴發的房顫相關[6];KONZEN等[7]報告了1例臨床及電生理缺乏肌強直表現的,最終經基因確診的DM1患者;有文獻研究結果表明男性的表現型比女性更嚴重[8];也有研究認為CTG擴張的中斷可能會導致更輕微的癥狀[9]。
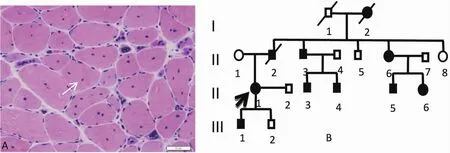
圖1 肌活檢HE染色可見大量核內移及核袋形成(A白色箭頭);患者的家系圖(B)調查患者家族18人中有10例伴有肌強直癥狀,其中2例已故(黑色箭頭為先證者)
關于DM1的初步診斷主要依據臨床癥狀:肌無力、肌萎縮、肌強直及電生理 (大量強直樣電位),肌肉活檢(明顯的核內移);最終確診需行基因檢測[10]。該患者臨床上缺乏肌無力及肌萎縮表現,肌強直癥狀起初未引起患者本身及臨床大夫的注意,加之患者伴有頭痛及認知功能障礙,行頭顱核磁顯示雙側大腦半球大致對稱皮層下白質病變,包括雙側顳極及右側外囊、額葉、頂葉及枕葉、雙側側腦室旁,影像學及臨床上疑似CADASIL診斷,后追問家族史,患者訴家族中有認知功能障礙及肌強直、白內障的家族史,家族中無腦卒中、偏頭痛病史,患者兒子發病較早,不符合CADASIL的發病年齡,給予患者肌電圖、肌活檢、基因檢測檢查后最終確診為DM1。
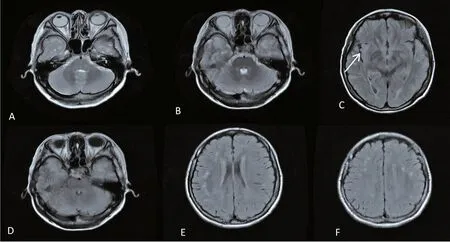
圖2 頭顱MRI雙側顳葉可見多發片狀長T2信號(A、B);右側外囊部位片狀高FLAIR信號(C白色箭頭),雙側額、頂、顳葉、側腦室旁大致對稱的高 FLAIR 信號(D、E、F)
CADASIL是一種與缺血性卒中和癡呆相關的遺傳性腦小血管病,其突變基因為Notch3。其最常見的臨床表現為缺血性小卒中、短暫性腦缺血發作、認知功能異常、偏頭痛等;本例患者影像學表現上疑似CADASIL,CADASIL頭顱核磁上常表現為雙側顳極白質和皮層下深部白質的病變,2001年O’SULLIVAN等[11]的研究認為核磁 T2上顳前葉的高信號是CADASIL的一種影像學標志,稱為O’Sullivan征,其對于CADASIL診斷的特異性和敏感性分別為90%和100%,研究認為其可能是由于擴大的血管周圍間隙(PVS)、髓鞘的退化以及缺乏組織間質液的引流導致。除此之外,因CADASIL是Notch3基因突變所致,其突變產物聚集在顱內小血管平滑肌,因此常表現為顱內的微出血、腦萎縮、腔隙性梗死灶、擴大的血管周間隙,當核磁伴有上述表現時應該考慮CADASIL的診斷[12],但灰質、白質的萎縮、腦室的擴大、擴張的血管周間隙常可出現于DM1患者中,并不具有特異性,而一些特征性的CADASIL核磁表現形式,例如在顳前葉[13]和外囊的長T2/FLAIR信號有時可出現在DM1中,因此當臨床上表現不典型時,單從影像學上鑒別較為困難;兩項意大利成人型DM1的系列研究顯示66%~80.4%的患者表現為白質異常,其中顳前葉白質異常為39~44%[14-15]。在HYUNJIN等[16]的研究中顯示顳前葉、頂枕葉、外囊、基底節區在DM1中的受累程度較CADASIL輕,另一方面外囊受累在DM1患者中發生率更低,大腦的微出血在DM1患者中沒有被發現,而在CADASIL中的發生率為69%,因此微出血可能是影像學上兩者的一個鑒別點;國內對于DM1顱內病變研究較少,其病變異質性較大,其中也常有伴有顱骨增厚的報告,其可能是DM1中樞性病變的一個影像學表現。
在DM1患者頭核磁上顳葉或腦室周圍白質異常的存在和嚴重程度與家族史和疾病持續時間有一定的聯系,與CTG重復大小沒有明確關聯,這些發現表明,其他的遺傳原因和/或未知的環境因素可能影響DM1患者病變的發生和嚴重程度[17]。
患有成人型DM1的患者會出現更多的大腦功能障礙,但是人們對神經癥狀的機制目前研究較少。有研究應用氫質子波譜成像(H-MRS)檢測到1/3的DM1患者腦脊液中伴有乳酸的異常積累,這些DM1患者與沒有乳酸積累的DM1患者相比,核磁上表現出相對較大的側腦室和較小的灰質體積,以及較重的白質病變。一些證據表明,tau蛋白的異常磷酸化可能會干擾線粒體的功能[18]。異常磷酸化的tau蛋白的積累和白質缺血性損傷的綜合作用可以解釋在一些DM1患者腦脊液中乳酸的異常積累。伴有乳酸聚集的DM1患者顯示出更高的白質病變和更嚴重的皮質萎縮,因此認為線粒體的功能異常在顱內病變中具有一定的作用。但是關于腦脊液乳酸水平、缺血性白質病變的嚴重程度和CTG擴張之間并沒有明確的關系[19]。研究認為能量代謝的缺陷可能在影響DM1患者的多器官病理過程中發揮作用,并為提高這些患者能量代謝的治療提供了理論依據[20]。
CADASIL與DM1在核磁共振成像上的表現有時可能是相似的,甚至可以表現出一些相似的癥狀,如認知功能障礙;關于DM1累及中樞性病變伴有認知功能障礙的國內研究較少,我們實驗室之前確診的DM1患者也未注意到中樞神經系統的病變[21],因此通過對本例患者的學習,我們要注重DM1患者顱內病變情況,尤其當臨床表現不典型,MRI上表現出雙側顳葉白質病變時,我們要想到DM1的可能,注重臨床查體與實驗室檢查及影像學、基因檢測的結合,以達到最終診斷,避免誤診。
