蛋白質二級結構在線服務器預測評估
2019-04-24 06:12:08朱樹平劉毅慧
生物信息學 2019年1期
朱樹平,劉毅慧
(齊魯工業大學(山東省科學院) 計算機科學與技術學院,濟南 250353)
蛋白質是人體的有機大分子,是生命活動的主要承擔者,在生物信息學領域,一直致力于對于蛋白質的研究。為了研究蛋白質的功能,往往從結構入手,但蛋白質結構有多種,其中關于二級結構的研究,有助于發現三維立體結構和提供蛋白質功能注解,因此大多數人都致力于蛋白質二級結構的研究。
在1951年,鮑林和科里首次提出了關于蛋白質二級結構問題[1],最初對于蛋白質二級結構的預測方法主要是通過研究氨基酸序列來進行,準確率在60%左右。Rost[2-3]等人在研究中采用PHD算法,把多序列排列中包含的進化信息作為神經網絡的輸入,預測蛋白質的二級結構準確率超過了70%。Zafer[4]等人使用動態貝葉斯分類器的稀疏算法,得到了76.3%的準確率。Kurniawan[5]等人使用SVM結合位置特異性打分矩陣(Position-specific scoring matrices,PSSM)和蛋白質結構的物理化學特征來預測,準確率達到80%左右。Wang[6]等人通過結合PSSM和氨基酸序列信息,并使用一種稱為二級結構遞歸編碼器-解碼器網絡(SSREDN)來解決輸入蛋白質特征與SS之間的序列-結構映射關系,使用CullPDB和CB513數據庫測試,分別達到84.2%,82.9%的Q3準確率。蛋白質二級結構預測方式不斷注入新的活力,現在很多方法都實現了在線服務器的預測,本文選取了PSRSM、MUFOLD、SPIDER、RAPTORX、JPRED和PSIPRED 6種服務器,分別闡述其算法原理,并通過測試數據比較每一個的預測準確度,從而給出當前在線服務器二級結構的評估。
1 在線服務器原理
1.1 PSRSM
該服務器使用基于數據分區和半隨機子空間(Partition and semi-random subspace method,PSRSM)的方法[7]。在傳統的隨機子空間方法中,低維子空間是由高維空間隨機采樣產生的,PSRSM使用的半隨機子空間方法能夠有效的保證基礎分類器的準確性和多樣化。該方法的主要步驟如下:首先把訓練數據根據蛋白質的長度劃分為不同的子集合,建立模型;然后使用半隨機子空間的方法生成子空間,并在子空間上訓練基礎分類器;最后根據多數投票的規則,在子集上把分類器結合起來,生成最終的分類器,其中使用SVM作為最基本的分類器。
具體來說,對于輸入使用PSI-BLAST程序生成PSSM數據,并且PSI-BLAST使用BLOSUM62進化矩陣搜索NCBI的非冗余(NR)數據庫的縮減版本,按照上述原則得到的PSSM是20*L的矩陣,20為氨基酸的個數,L為每個蛋白質的長度。在實驗中使用13個滑動窗口來獲取蛋白質序列信息和預測序列中心的蛋白質二級結構。假設輸入一個長度為L的蛋白質,會產生260*L(13*20*L)的輸入矩陣。從260個特征值選取160個作為主要特征,作為網絡輸入。最后建立12個分類器進行訓練。那么一個新的蛋白質序列會根據其長度,選擇合適的分類器進行預測。
實驗的訓練集選取了ASTRAL數據集的6 892條蛋白質數據和CullPDB數據集的12 288條蛋白質數據,去掉相似度較高的蛋白質后,訓練集總共包括15 696條數據。測試集使用99個CASP10數據、81個CASP11數據、19個CASP12數據、513個CB513數據、1 673個25PDB的數據和2018年2月1號之前的100條數據(T100),實驗得到使用6個GTPCs模型在25PDB、CB513、CASP10、CASP11、CASP12和T100數據中的蛋白質二級結構的Q3預測準確率分別是86.38%、84.53%、85.51%、85.89%、85.55%和85.09%。該服務器預測蛋白質序列范圍是10到800,預測網址為:http://qilubio.qlu.edu.cn:82/protein_PSRSM/default.aspx。
1.2 MUFOLD
MUFOLD采用的是一種名為深度初始-內部-初始(Deep 3I)的新型網絡來預測蛋白質二級結構,并且對于輸入的特征矩陣做了細致考量,特征矩陣中結合了氨基酸的理化性質、PSI-Blast特征和HHBlits特征[8]。其中對于理化性質的特征矩陣,設置了從-1到1之間選取的8個數字來表示一個氨基酸,前7位表示氨基酸理化性質,后一位用1或0表示是否輸入氨基酸。如表1,“*”表示某一類氨基酸,“n”表示依據理化性質設置的數值。MUFOLD設置默認輸入矩陣為700*8,若假設輸入一個氨基酸序列個數為600的蛋白質,設置矩陣時會把前600行的前7位按照本身理化性質設置,第8位設為0,而后100行的前7為全部設為0,后一位設置為1。
對于PSI-Blast的特征,按照類似原理用從0到1的選取21位數字表示一個氨基酸,前20位根據得到的PSSM值設置,后一位用1或0表示是否有輸入;對于HHBlits特征則用0到1之間的31位數字表示一個氨基酸,前30位根據HMM文件設置,最后一位同樣用0或1表示輸入。以上三個特征被組合成一個58位的特征,作為網絡的輸入。
Deep3I網絡是由2個Deep3I塊、一系列卷積和完全聯通的致密層構成。而Deep3I塊是由初始模塊遞歸嵌套構成,初始模塊通過卷積操作能夠有效提取氨基酸殘基之間的非局部相互作用。Deep3I網絡通過用TensorFlow和Keras不斷進行訓練和實驗來對蛋白質二級結構進行預測。
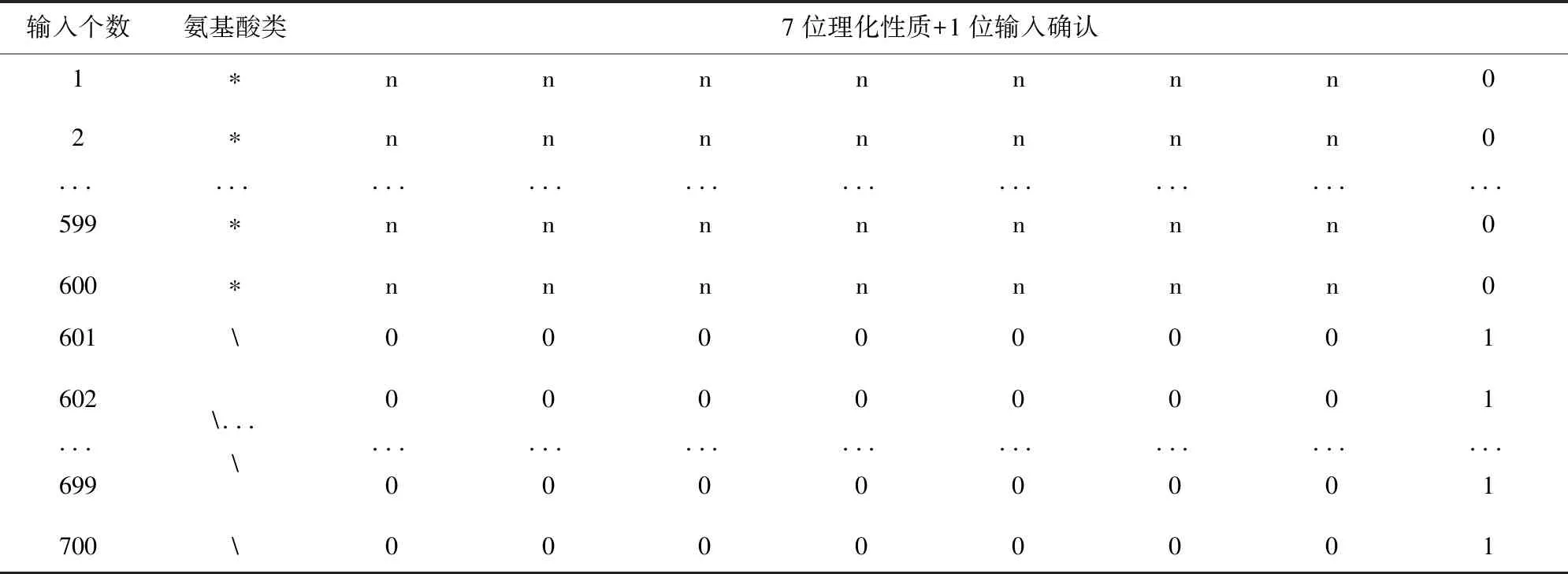
表1 按照氨基酸理化性質設置的輸入矩陣Table 1 Input matrix set according to the physical and chemical properties of amino acids
MUFOLD實驗中的數據集使用蛋白質序列長度介于50到700之間的數據,來自CullPDB、JPRED、CASP、CB513和PDB 5個公開的蛋白質數據庫。具體來說:從CullPDB選取了9 581條數據,其中隨機選出9 000條作為訓練集,剩下的581條作為測試;從JPRED選取的數據均來自不同的超級家族;CASP的數據集經過篩選后CASP10的98條數據,CASP11的83條數據,CASP12的40條數據被使用;CB513和385條PDB數據也同樣被應用于MUFOLD的實驗中。MUFOLD測試數據的范圍是30到700,測試網址是:http://mufold.org/mufold-ss-angle/。
1.3 SPIDER
Hefferman[9]等人提到對于蛋白質二級結構預測和溶劑接觸表面積的研究,多年一直停滯不前的原因來自于,有些氨基酸殘基在三維結構中距離很近而在蛋白質序列中距離很遠,因此較難捕獲氨基酸殘基之間的非局部相互作用。現有的機器學習的方法基本都使用10~20個滑動窗口來獲取氨基酸的相互作用。而SPIDER不使用滑動窗口,采用一種長期短期記憶(Long Short-Term Memory , LSTM)雙向遞歸神經網絡(Bidirectional Recurrent Neural Networks ,BRNNs)的機器學習模型來實現預測,能夠捕捉氨基酸殘基之間的非局部相互相互作用,實驗證明它能夠改善蛋白質二級結構、骨干角度、接觸號碼和溶劑可及性的預測。
該網絡的LSTM-BRNN模型是由兩個使用LSTM細胞的BRNN層和兩個緊密連接用整流線性單元(Rectified Linear Unit, ReLU)激活的隱含層構成,它被用于四次迭代中。對于該網絡的輸入,包含了7種具有代表性的蛋白質氨基酸理化性質(Physio-chemical properties,PP)、20維來自PSI-Blast的PSSM和30維來自HHBlits每個殘基的隱藏馬爾科夫模型的序列譜(HMM Profiles),把這些數據放入由LSTM-BRNNs網絡構成的迭代中,進行四次迭代(其中一次迭代包括兩個LSTM-BRNN),最后得到最終機器學習模型。該過程主要結構如圖1所示。在訓練期間為防止過擬合,使用丟失率為50%的丟失算法,并用Adam優化訓練過程,該網絡能夠在不使用滑動窗口的條件下捕獲長短距離交互。
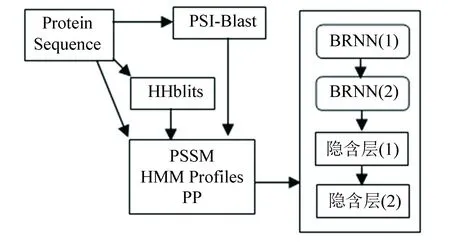
圖1 SPIDER 主要結構Fig.1 Main structure of SPIDER
1.4 RAPTORX
RAPTORX使用由深度卷積神經網絡(Deep convolutional neural network , DCNN)和條件隨機場(Conditional random fields,CRF)組合而成的深度卷積神經場(Deep Convolutional Neural Fields,DCNF),來預測蛋白質二級結構,并且對網絡采用一種在ROC曲線下面積的(Area under the ROC curve,AUC)最大化方法來訓練,從而能夠很好地解決紊亂序列蛋白質的預測問題[11]。Wang[12]提到在使用蛋白質序列文件后,RAPTORX在數據集CASP和CAMEO能夠得到大約為84%的Q3準確率和72%的Q8準確率,不使用序列文件能夠獲得約為74%的Q3準確率和59%的Q8準確率,它能夠有效的解決復雜的基因結構關系建模和相鄰殘基間的建模。Wang[13]指出DCNF使用DCNN代替CNF中使用的淺層神經網絡,能夠捕獲輸入和輸出標簽之間復雜的關系,并且能夠捕獲遠程的序列信息。
RAPTORX實驗中使用的數據有6 125個CullPDB數據,CB513數據、123個CASP10數據、105個CASP11數據和CAMEO的數據,還有JPRED公開的1 338個訓練數據和149個測試數據。RAPTORX測試數據范圍是26到4 000個蛋白質序列,預測網址為:http://raptorx.uchicago.edu/StructurePropertyPred/predict/。
1.5 JPRED
JPRED服務器從1998年開始提供蛋白質的預測到現在已經發展到JPRED4版本。JPRED3版本用JNET算法提供單個蛋白質序列或者多序列比對(MSA)的預測,其中JNET使用JNET v2.0。JNET v2.0不使用頻率文件,只使用PSI-BLAST的PSSM配置文件和HMMER的隱馬爾可夫模型,把神經網絡由9個單元增加到100個單元,該方法是通過對超家族級別的SCOPe數據的Astral匯編衍生的序列和結構非冗余數據集進行7倍交叉驗證培訓而開發的[14],最后使用149條盲數據進行測試得到了81.5%的Q3準確率。
JPRED4版本和JPRED3一樣,同樣使用JNET算法并提供單一序列和多序列比對的蛋白質序列的二級預測。不同的是它選取1 358個SCOPe/ASTRAL v.2.04 超級家族中的一個為代表,用JNET 2.3.1進行7倍交叉驗證的實驗,通過尋找UniRef90 v.2014_07來生成PSI-BLAST文件并為每一個蛋白質序列建立多重序列比對。最后在150個訓練集上獲得了82%的準確率[15]。同時JPRED在線服務器也可以提供溶劑可及性和卷曲螺旋區的預測,預測網址為:http://www.compbio.dundee.ac.uk/jpred4/index.html。
1.6 PSIPRED
Mcguffin[16]等人指出PSIPRED服務器結合了三種先進的技術,分別是PSIPRED、GenTHREADER和MEMSAT 2。其中PSIPRED采用嚴格的交叉驗證程評估性能,并且采用兩個前饋的神經網絡,對從PSI-BLAST獲得的輸出進行分析,從而得到可靠的二級結構預測結果;GenTHREADER用來推斷跨膜蛋白的結構和拓撲結構;MEMSAT2能夠快速識別蛋白質的折疊信息,預測網址為:http://bioinf.cs.ucl.ac.uk/psipred/。
從以上6個服務器預測過程的角度分析,可以看到每個服務器各有優缺點。其中能夠批量上傳和下載實驗結果的是PSRSM、SPIDER3和RAPTORX,給定結果為壓縮包的形式,需進一步整合。服務器JPRED和PSIPRED都必須遵循每次只能上傳一個蛋白質文件(或序列)的約定,而且結果是以郵件的形式發送到郵箱里面,并且PSIPRED在同一時間段內最多只允許上傳20條數據進行預測,因此預測結果獲取過程較為復雜。MUFOLD雖然網站上說明一次可以批量上傳少于10條的數據但是在實驗中獲取數據,最多一次只可上傳4條數據進行預測。6個服務器預測的時間相差并不是很大,主要在于預測結果的獲取方式上存在很大差距。
2 數據選取和評估標準
基于每個服務器都可以預測為前提,依據蛋白質發布的月份和其同源性分別選取了150條數據進行實驗,并采用了合適的評價標準來評估。
2.1 數據選取
數據選取遵循以下原則:數據選取2018年PDB最新發布的數據,保證了測試集不在服務器的訓練集中;數據來自不同的時間段,更具有分散性;數據量較大,使得實驗結果更具有說服力;選取的蛋白質長度能夠讓每一個服務器都可以進行測試,并得到預測結果。基于上述的條件從2018年4、5、6月份分別選取了50條蛋白質序列進行第一次實驗,數據選取如表2所示。
并且為了使實驗結果更具有可靠性,又進一步從2018年4到8月,基于同源性的30%,50%和70%隨機分別選取了50條數據,共150條數據(T150)進行第二次實驗,該實驗的數據選取如表3所示。
2.2 評估標準
本文采用了兩種衡量蛋白質二級結構預測準確性方法:Q3和Sov的值主要是衡量個別殘基分配的精度,Sov的值主要是衡量全元素的預測精度。
2.2.1Q3
按照DSSP[17]的規定,通常我們把蛋白質二級結構劃分為H、G、I、E、B、T、S和-,8種狀態。而這8這種狀態,按照H、G、I→H,E、B→E,其他→C的方式,將一條氨基酸序列轉化為H(螺旋)、E(折疊)、C(卷曲),3種狀態。則Q3表示被正確預測的三種狀態的氨基酸數占整個氨基酸序列的比例。符合以下計算公式:
(1)
其中:SE是E類蛋白質結構準確預測的數量,SH是H類蛋白質結構準確預測的數量,SC是C類蛋白質結構準確預測的數量,S是指總的氨基酸數量,Q3指的是三種狀態下,蛋白質二級結構預測的準確率。

表2 DB150 數據集Table 2 DB150 data set

表3 T150 數據集Table 3 T150 data set
2.2.2Sov
Sov的計算是基于重疊片段比值的一種測度,它對預測結果和觀察到的結果同等對待。同樣按照上述Q3的思想把蛋白質二級結構劃分為螺旋、折疊和卷曲三種狀態。如果假設觀察到的序列記為S1,預測到的序列記為S2,S0為S1和S2所有狀態相同的片段,那么S0必定會包含一對重疊和一個螺旋,接下來S1的長度為length(S1),并且把每對中S1和S2序列個數求并集記為max(S1,S2),把S1和S2的序列個數求交集記為min(S1,S2)。在上述基礎上把Sov的計算公式定義為[18]:
(2)
其中關于δ的設定是為了允許蛋白質結構中邊緣處片段的變化,δ(S1,S2)取值符合以下定義:
(3)
3 實驗及結果
從PDB中下載得到最新的蛋白質數據,然后分別上傳到6個預測服務器上進行測試。上傳蛋白質序列得到的預測結果后,通過與正確的三態的DSSP結果相比較,計算每一條蛋白質的Q3和Sov準確率。第一次實驗中每月數據和DB150的Q3和Sov準確率如表4所示。第二次實驗中基于30%,50%,70%的同源度數據和T150的Q3和Sov的實驗結果如表5所示。
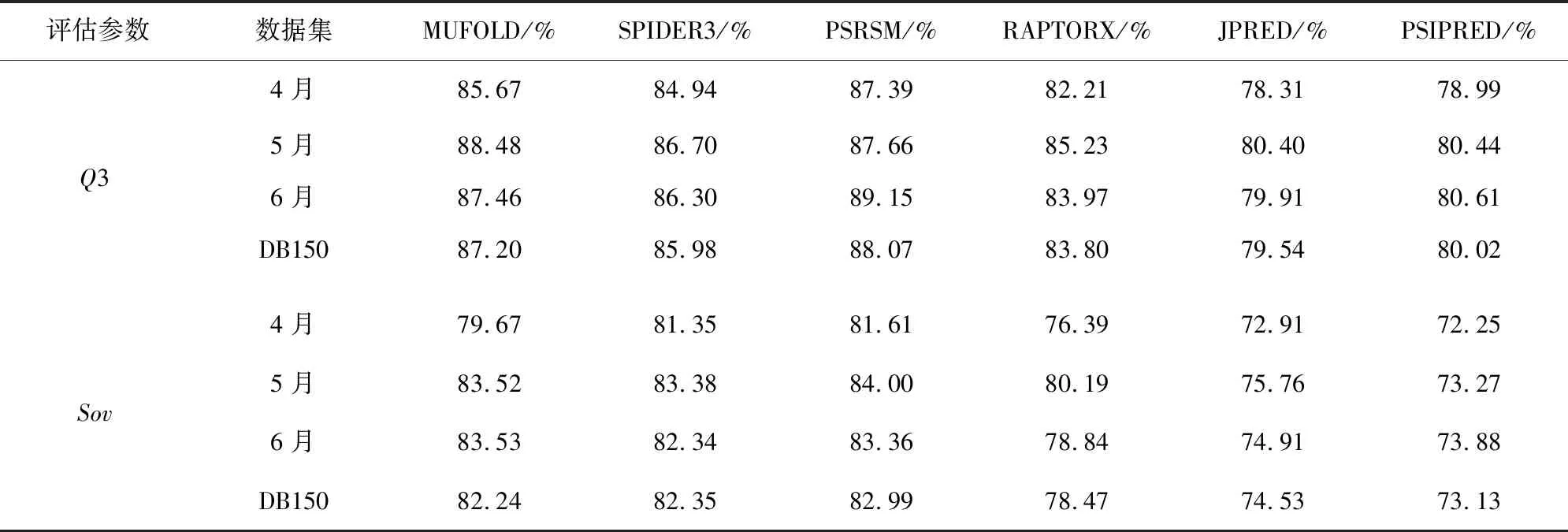
表4 實驗1的Q3和 Sov平均準確率Table 4 Average accuracy of Q3 and Sov in Experiment 1
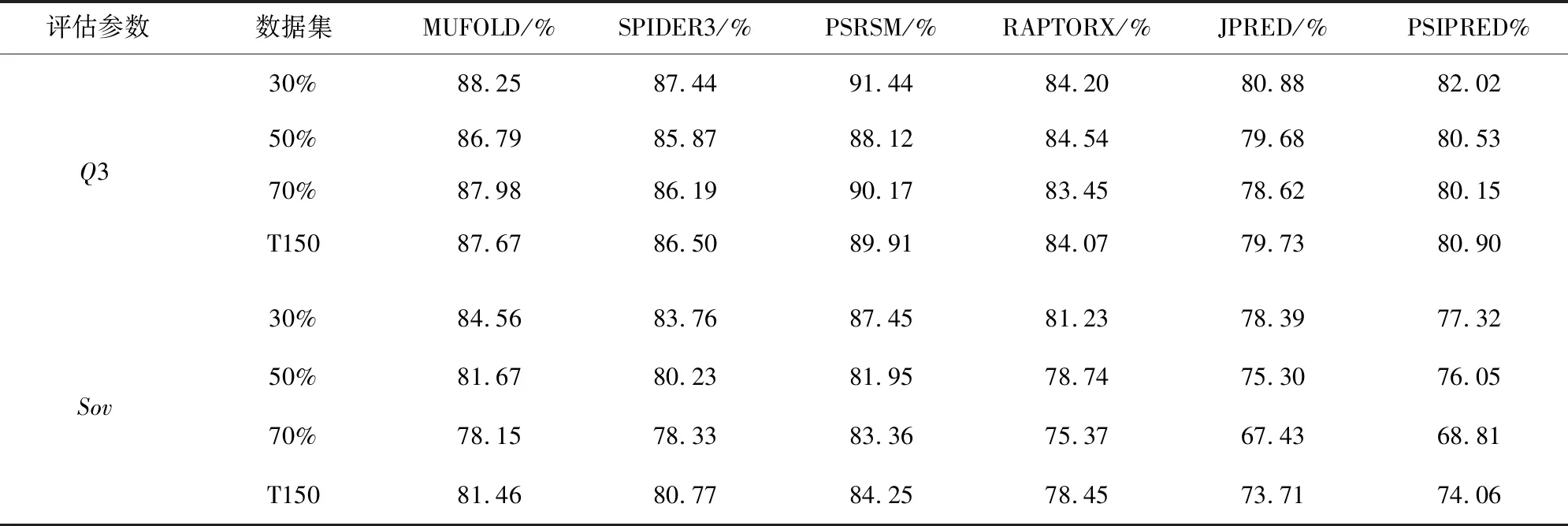
表5 實驗2的Q3和Sov平均準確率Table 5 Average accuracy of Q3 and Sov in Experiment 2
從實驗結果中看到,不論是基于月份的蛋白質數據,還是基于同源性不同劃分的數據,PSRSM都取得了在同一類別中較好的效果,Q3的預測準確率有時甚至超過90%。按照月份劃分時,4月份的數據集中,PSRSM達到了最好的預測效果,Q3和Sov的值分別為87.39%和81.61%;在5月份數據集中,MUFOLD的Q3準確率最高,為88.48%,Sov準確率僅次于PSRSM的84.00%,為83.52%;在6月份數據集中PSRSM的Q3獲得最高準確率為89.15%,而Sov僅次于MUFOLD的83.53%,為83.36%。在綜合數據DB150的結果中我們得到6種預測方式Q3的準確率由高到低為PSRSM的88.07%,MUFOLD的87.20%,SPIDER的85.98%,RAPTORX的83.80%,PSIPRED的80.02%和JPRED的79.54%;Sov準確率由高到低為PSRSM的82.99%,SPIDER3的82.35%,RAPTORX的78.47%,JPRED的74.53%和PSIPRED的73.13%,PSRSM得到了Q3和Sov的最高準確率。
在基于同源性的實驗中,結果顯示基于30%時,PSRSM得到了91.44%的Q3準確度和87.45%的Sov準確度,比其他服務器中最好的MUFOLD分別高出3.19和2.89個百分分點;同源度為50%時,PSRSM的Q3為88.12%,Sov為81.95%,分別比MUFOLD高出1.33和0.28個百分點;70%的同源度時PSRSM的Q3和Sov分別為90.17%和83.36,Q3比其他服務器中最好的MUFOLD高出2.19%,Sov比預測結果最好的SPIDER高出5%。總體來看在T150中Q3和Sov準確率由高到低分別為PSRSM的89.91和84.25%,MUFOLD的87.67%和81.46%,SPIDER的86.50%和80.77%,Raptorx的84.07%和78.45%,PSIPRED的80.06%和74.06%,JPRED的79.73和73.71%。
無論在哪一種情況下,PSRSM、MUFOLD和SPIDER3都得到了超過84.9%的Q3準確率和超過78.1%的Sov準確率,其中PSRSM表現出良好的預測性能。
4 結 論
蛋白質二級結構預測的準確度,將決定人類對于蛋白質功能的了解程度。本文介紹了現在6個熱門的預測服務器原理,并使用最新的數據對其二級結構預測的準確率進行評估。比較6個服務器的預測方法和實驗結果,可以看到它們的研究方法都在著重解決那些三維結構中距離近而序列中距離遠的氨基酸殘基的預測問題,并為此一再提出新的解決思路。
PSRSM在上述實驗數據中大多都取得了最好的實驗結果,特別是在基于同源性差異的實驗中,當同源度較很低為30%時,其Q3準確率比其他服務器中最好的MUFOLD高出3.19%,這更說明PSRSM具有更好的預測效果。PSRSM與其他服務器比較,其優點在于基于蛋白質長度劃分設計模板的使用,另一點在于訓練數據量非常龐大,當然也采用了合理的預測方法。通過該實驗和結果也可以看出,其他服務器能否獲得優越的結果與其訓練數據量的大小密切相關,當然還與其各自使用的深度學習算法有關。因此今后對于蛋白質二級結構預測的研究應當重點從大數據、模板和深度學習的角度進行突破。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
現代企業(2015年9期)2015-02-28 18:56:50
