基于機(jī)器學(xué)習(xí)的RNA編輯位點(diǎn)預(yù)測方法綜述
2019-04-24 06:12:06冷嘉承吳凌云
生物信息學(xué) 2019年1期
關(guān)鍵詞:方法
冷嘉承,吳凌云*
(1.中國科學(xué)院數(shù)學(xué)與系統(tǒng)科學(xué)研究院 應(yīng)用數(shù)學(xué)研究所,管理、決策與信息系統(tǒng)重點(diǎn)實(shí)驗(yàn)室,國家數(shù)學(xué)與交叉科學(xué)中心,北京 100190; 2.中國科學(xué)院大學(xué) 數(shù)學(xué)科學(xué)學(xué)院,北京100049)
RNA合成、加工、行使功能和降解是細(xì)胞生存的關(guān)鍵,并在許多不同的層面進(jìn)行著調(diào)控[1]。RNA合成是基因表達(dá)的第一步,轉(zhuǎn)錄因子調(diào)控RNA聚合酶II(Pol II)與啟動(dòng)子結(jié)合[2],通過一套非常復(fù)雜的操作步驟將DNA轉(zhuǎn)錄為前體RNA[3]。前體RNA隨后被加工產(chǎn)生成熟mRNA、功能性tRNA和rRNA[4]。RNA的加工包括(1)加帽:將7-甲基鳥苷酸(m7G)添加到5’末端[5];(2)聚腺苷酸化:在3’末端添加poly-A尾巴[6];(3)剪接:去除內(nèi)含子之后拼接外顯子[7];(4)RNA編輯:修改RNA分子序列并導(dǎo)致蛋白質(zhì)多樣性[4, 8]。而RNA編輯作為RNA加工中的一環(huán),起著至關(guān)重要的作用。
RNA編輯有很多種改變RNA序列的機(jī)制,其中涉及了堿基的插入和缺失以及堿基替換,如胞苷(C)到尿苷(U)和腺苷(A)到肌苷(I)的脫氨基化,以及非模板化的核苷酸添加和插入[3]。到目前為止,人們已經(jīng)在真核生物、病毒、古細(xì)菌以及原核生物中發(fā)現(xiàn)了RNA編輯。所以,生物學(xué)家一直對RNA編輯保持著廣泛的關(guān)注和強(qiáng)烈的興趣[1-3, 8]。
在哺乳動(dòng)物中已經(jīng)發(fā)現(xiàn)了兩種類型的RNA編輯。一種是由催化性類多肽載脂蛋白B mRNA編輯酶(APOBEC)催化的C至U編輯,其頻率相當(dāng)?shù)汀A硪环N是A-to-I編輯,其中腺苷通過腺苷脫氨酶(ADAR)的作用脫氨基成肌苷,其頻率非常高。因?yàn)槿祟怰NA編輯中絕大多數(shù)的編輯都屬于A-to-I編輯,故A-to-I編輯在生物細(xì)胞分子機(jī)制中尤為重要。研究表明,A-to-I RNA編輯同時(shí)也與腦功能、病毒感染和人類疾病有關(guān)。例如在人體中,RNA合成和加工中的錯(cuò)誤可能引起神經(jīng)系統(tǒng)疾病,如三核苷酸擴(kuò)張性疾病強(qiáng)直性肌營養(yǎng)不良和脆性X綜合征[8]。這些均顯現(xiàn)了RNA編輯研究的重要性與意義。
深度測序技術(shù)和生物信息學(xué)的發(fā)展使得人們可以在全局篩選A-to-I RNA編輯位點(diǎn)。但是目前為止,通過高通量測序數(shù)據(jù)準(zhǔn)確識(shí)別RNA編輯事件仍然是一個(gè)巨大的挑戰(zhàn)。現(xiàn)在的方法通常將短讀段映射到參考基因組或轉(zhuǎn)錄組,然后去除相同的讀段,過濾低質(zhì)量讀段,調(diào)用差異信息并且去除已知的單核苷酸多態(tài)性(SNP)[6, 7, 10, 15-17]。但是將大量的短讀段映射到參考基因組是非常耗時(shí)的,并且只有少數(shù)流水線和計(jì)算工具可公開用于處理RNA編輯[18]。最關(guān)鍵的是,由于成本問題,DNA測序與RNA測序一般不會(huì)一起進(jìn)行,所以很難區(qū)分新的SNP位點(diǎn)和RNA編輯位點(diǎn)。實(shí)際上,大量的RNA編輯位點(diǎn)已被注釋為dbSNP中的SNP[19]。
針對這些現(xiàn)象,尤其是為了區(qū)分SNP位點(diǎn)和RNA編輯位點(diǎn),陸續(xù)產(chǎn)生了許多能夠較為準(zhǔn)確預(yù)測RNA編輯位點(diǎn)的方法,本文對一些常見方法進(jìn)行了總結(jié)(見表1),將在第三節(jié)對這些方法進(jìn)行詳細(xì)的介紹。
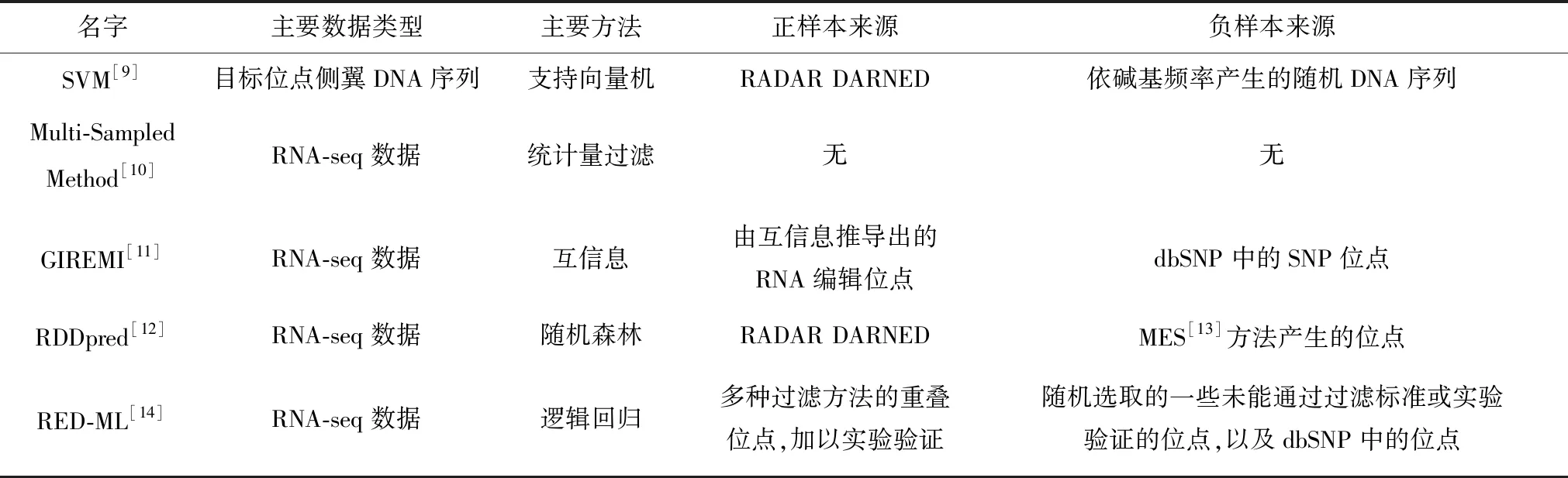
表1 RNA編輯位點(diǎn)預(yù)測方法概覽Table 1 Overview of RNA editing site prediction methods
注:正樣本指發(fā)生RNA編輯的位點(diǎn)(包括所有類型的RNA編輯,以A-to-I編輯為主),負(fù)樣本指發(fā)生堿基變化但沒發(fā)生RNA編輯的位點(diǎn),其中SVM方法只針對A-to-I編輯。
1 RNA編輯數(shù)據(jù)庫
已有文獻(xiàn)在研究和評價(jià)RNA編輯位點(diǎn)預(yù)測方法時(shí)使用了不同的數(shù)據(jù)集,目前還沒有一個(gè)通用的、被廣泛接受的基準(zhǔn)數(shù)據(jù)集。除了一些文獻(xiàn)中提供的獨(dú)立數(shù)據(jù)集,隨著越來越多的RNA編輯位點(diǎn)被發(fā)現(xiàn),目前已經(jīng)出現(xiàn)了多個(gè)關(guān)于RNA編輯的數(shù)據(jù)庫。這些數(shù)據(jù)庫為研究基于計(jì)算的RNA編輯位點(diǎn)預(yù)測方法提供了很好的訓(xùn)練數(shù)據(jù)和研究的基礎(chǔ)。
1.1 RADAR數(shù)據(jù)庫
Ramaswami和Li等人于2013年創(chuàng)建了RADAR數(shù)據(jù)庫[20],包括了人類、蒼蠅、老鼠這三個(gè)物種(hg19/mm9/dm3)的A-to-I RNA編輯數(shù)據(jù)。該數(shù)據(jù)庫可以根據(jù)用戶的要求進(jìn)行篩選,例如基因名稱、是否位于Alu區(qū)域、與其他物種之間的編輯保守性等,并且提供了一些常用數(shù)據(jù)的直接下載,如hg19中的全部RNA編輯位點(diǎn)等。在結(jié)果中點(diǎn)擊位置信息可以鏈接到UCSC基因組瀏覽器上瀏覽更詳細(xì)的信息,并且都附有數(shù)據(jù)的文獻(xiàn)來源。
1.2 DARNED數(shù)據(jù)庫
DARNED數(shù)據(jù)庫[21]是由Kiran和Baranov創(chuàng)建的。它包括多種數(shù)據(jù)來源:(1)生物信息學(xué)分析cDNA序列和基因組序列之間的差異;(2)SNP的分析數(shù)據(jù);(3)miRNA的分析數(shù)據(jù);(4)來自同一組織的RNA和DNA樣品的高通量測序結(jié)果[21]。最后將RNA編輯事件的位點(diǎn)映射到參考人類基因組。此數(shù)據(jù)庫不僅包括RADAR中的三個(gè)基因組還包括了hg18以及mm10。目前為止該數(shù)據(jù)庫支持三種RNA編輯位點(diǎn)的查詢方式:根據(jù)區(qū)域(染色體、位置、組織特異性等信息)查詢;根據(jù)基因名稱(如APOB)查詢;根據(jù)序列本身查詢。在此數(shù)據(jù)庫的查詢結(jié)果中,通過點(diǎn)擊位置信息不僅可以鏈接到UCSC上,還可以鏈接到ENSEMBL上,比較方便。對是否與SNP混淆,與哪個(gè)SNP混淆,基因名稱,基因區(qū)域都有詳細(xì)的標(biāo)注,而且都可以通過點(diǎn)擊基因信息鏈接到NCBI上進(jìn)行查詢,十分人性化。
2 RNA編輯位點(diǎn)預(yù)測方法
2.1 基于DNA序列數(shù)據(jù)的有監(jiān)督學(xué)習(xí)方法
Sun等人[9]提出了一個(gè)支持向量機(jī)(SVM)模型,基于DNA測序數(shù)據(jù)對A-to-I類型的RNA編輯位點(diǎn)進(jìn)行預(yù)測。其核心思想是,RNA編輯位點(diǎn)主要是由該位點(diǎn)附近的序列決定的。該方法將序列與序列之間的關(guān)系進(jìn)行相關(guān)性分析,得到相似矩陣,然后通過映射將其轉(zhuǎn)化到內(nèi)核空間得到核矩陣,最終利用SVM模型進(jìn)行訓(xùn)練,得到具有判別能力的RNA編輯位點(diǎn)分類器(見圖1)。
序列間的相關(guān)性是由字符串距離刻畫的:
(1)
其中a,b分別代表輸入的兩個(gè)字符串,DEdit代表的是編輯距離,即a,b之間互相轉(zhuǎn)換最少需要插入、刪除或者替換多少個(gè)字符,而DHamming代表的是漢明距離,與編輯距離不同,只允許替換。而L1,L2分別代表計(jì)算編輯距離和漢明距離所用的字符串長度(見圖1)。w是0到1之間取值的權(quán)重。D的值越大,代表兩個(gè)序列之間的相似性越低。該方法在LIBSVM[22]中使用字符串核函數(shù)將字符串?dāng)?shù)據(jù)轉(zhuǎn)換為向量空間。字符串核函數(shù)是對字符串類型數(shù)據(jù)進(jìn)行操作的核函數(shù),可寫為:
K(a,b)=exp(-gamma×D(a,b)2)
(2)
其中D是從等式(1)導(dǎo)出的組合距離。伽瑪參數(shù)定義單個(gè)訓(xùn)練實(shí)例影響到達(dá)的距離,低值表示“遠(yuǎn)”,高值表示“近”。
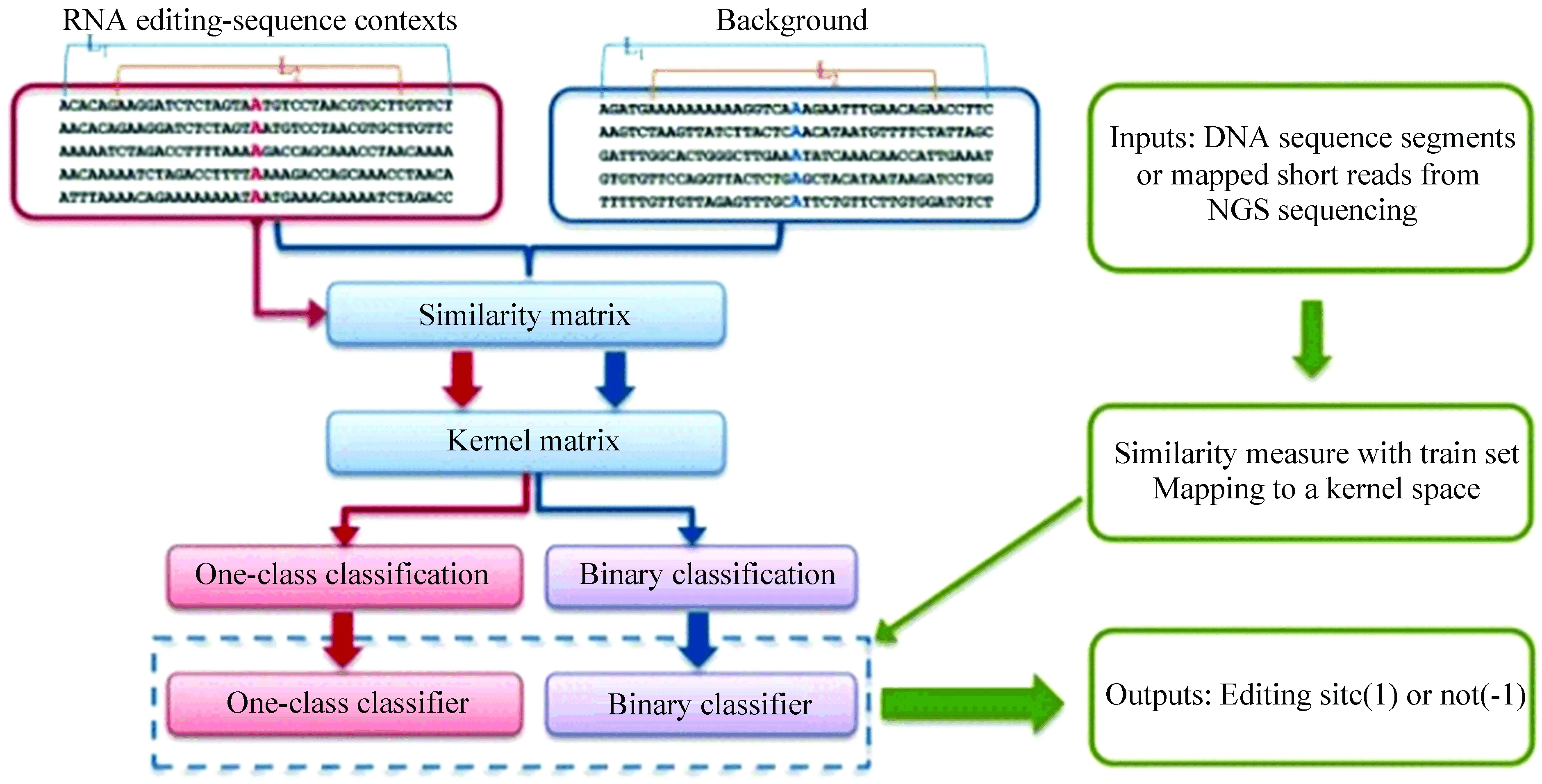
圖1 SVM方法流程圖[9]Fig.1 Flow chart of SVM method[9]
該論文還嘗試了單類SVM。與傳統(tǒng)的支持向量機(jī)相比,單類支持向量機(jī)嘗試學(xué)習(xí)一個(gè)決策邊界,實(shí)現(xiàn)樣本與原點(diǎn)之間的最大分離。他們根據(jù)Sch?lkopf等人的研究結(jié)果[23],使用二進(jìn)制值1作為編輯事件,-1作為非編輯事件。該文采用5折交叉驗(yàn)證的方式得到,僅用正樣本作為訓(xùn)練數(shù)據(jù)的單類SVM的性能要遠(yuǎn)低于雙類SVM,單類SVM在參數(shù)nu=0.5時(shí)的精確度在果蠅、小鼠、人類數(shù)據(jù)集上分別是0.489, 0.495, 0.498,而雙類SVM模型卻分別達(dá)到了0.75, 0.85, 0.79左右。并且單類SVM方法通常無法權(quán)衡在正負(fù)樣本上性能的差異。該文還用基于人類樣本訓(xùn)練的模型在一個(gè)Sanger測序集上進(jìn)行了驗(yàn)證試驗(yàn),其中79.3%(46/58)的位點(diǎn)被成功預(yù)測。
2.2 基于RNA-seq數(shù)據(jù)的無監(jiān)督學(xué)習(xí)方法
僅僅基于DNA序列的預(yù)測方法需要可靠的RNA編輯位點(diǎn)數(shù)據(jù)作為訓(xùn)練樣本,而且這類方法對于訓(xùn)練樣本的依賴性非常高。考慮到不同物種和不同類型樣本中發(fā)生RNA編輯概率的差異,以及準(zhǔn)確的RNA編輯位點(diǎn)數(shù)據(jù)的缺乏,目前更為常見的是從RNA-seq數(shù)據(jù)中識(shí)別出RNA編輯位點(diǎn)。按照使用的機(jī)器學(xué)習(xí)方法的不同,基于RNA-seq的RNA編輯位點(diǎn)識(shí)別方法又可以大致分為兩類:基于無監(jiān)督學(xué)習(xí)的過濾方法,和基于有監(jiān)督學(xué)習(xí)的機(jī)器學(xué)習(xí)方法。第一類方法通過比較RNA-seq數(shù)據(jù)和參考基因組的差異,獲得了潛在的RNA編輯位點(diǎn),然后通過與SNP數(shù)據(jù)庫的比較以及其他指標(biāo)進(jìn)行過濾,去除假陽性位點(diǎn),最終獲得較為可靠的RNA編輯位點(diǎn)。而第二種方法則通過RNA-seq數(shù)據(jù)和參考基因組提取出一些特征,建立機(jī)器學(xué)習(xí)模型,利用已知的RNA編輯位點(diǎn)數(shù)據(jù)進(jìn)行有監(jiān)督訓(xùn)練,獲得RNA編輯位點(diǎn)的預(yù)測模型。
Ramaswami等人[10]提出了整合多樣本RNA-seq數(shù)據(jù)識(shí)別RNA編輯位點(diǎn)的方法。為了利用大量可公開獲得的RNA-seq數(shù)據(jù)集來發(fā)現(xiàn)RNA編輯位點(diǎn),該文章提出了兩種相關(guān)且互補(bǔ)的方法,以使用來自單個(gè)物種中的多個(gè)個(gè)體的RNA-seq數(shù)據(jù)準(zhǔn)確鑒定RNA編輯位點(diǎn)。在第一種方法(見圖2(a))中,在每個(gè)RNA-seq樣品中分別將測序讀段映射到(非匹配的)基因組參考序列后,找出RNA的變化,并且將已知常見基因組SNP去除,以此將RNA編輯位點(diǎn)與其余稀有SNP區(qū)分開來。這主要是因?yàn)橄嗤木庉嬑稽c(diǎn)通常存在于不同的個(gè)體中,而罕見的SNP很可能不存在。
在第二種方法中(見圖2(b)),將不同樣本的RNA-seq的讀段匯總到一起進(jìn)行比對,從而提高找出RNA變異的靈敏度。然后如第一種方法一樣,按接下來的步驟排除掉SNP,并且找出RNA編輯。由于罕見的SNP不可能出現(xiàn)在多個(gè)個(gè)體中,所以在匯聚后的比對中將以非常低的頻率存在。

圖2 Multi-Sampled Method的兩種方法[10]Fig.2 Two methods of Multi-Sampled Method[10]
而GIREMI方法[11]則是典型的僅需要單組RNA-seq的方法,GIREMI將RNA-seq讀段中單核苷酸變異(SNV)對的統(tǒng)計(jì)推斷模型與機(jī)器學(xué)習(xí)方法結(jié)合,以預(yù)測RNA編輯位點(diǎn)。GIREMI的輸入包括來自RNA-seq數(shù)據(jù)集的SNV(錯(cuò)配)列表和公共數(shù)據(jù)庫(如dbSNP)中已知的SNP,輸出是預(yù)測的RNA編輯位點(diǎn)及其編輯水平。除了公開的SNP信息外,GIREMI僅僅使用感興趣的RNA-seq數(shù)據(jù)集進(jìn)行所有分析,而不依賴于任何其他基因組或RNA-seq數(shù)據(jù)集,因此這種方法適用于更廣的范圍(見圖3)。
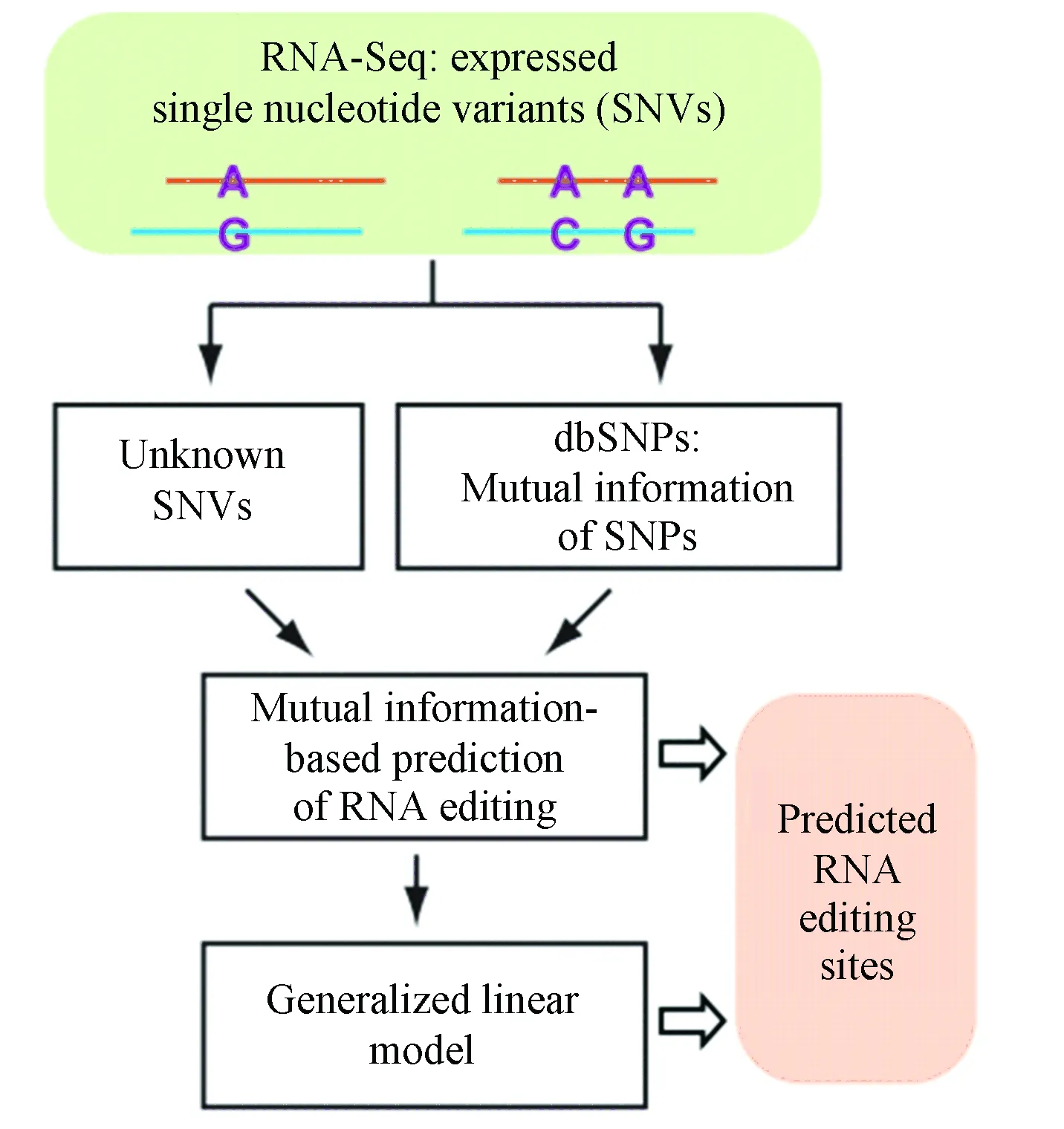
圖3 GIREMI流程圖[11]Fig.3 Flow chart of GIREMI[11]
GIREMI分為兩個(gè)步驟,首先進(jìn)行SNV位點(diǎn)的互信息計(jì)算:
對于每個(gè)SNV,我們考慮所有可能的堿基A,T,C,G作為變量si的四種可能狀態(tài)。對于表示一對堿基(si,sj)的聯(lián)合變量,總共有16種可能狀態(tài)。各種狀態(tài)si,sj或(si,sj)的概率可以使用最大似然法進(jìn)行估計(jì)得到。考慮到所有可能的測序錯(cuò)誤以及實(shí)際數(shù)據(jù)中的低測序深度,假設(shè)在實(shí)際數(shù)據(jù)中未觀察到的狀態(tài)的概率值為0.01。因此(si,sj)的互信息是:
(3)
其中N={A,T,C,G},ni和nj分別表示si和sj的狀態(tài)。而SNPsi的信息值被定義為:
(4)
其中S代表帶有si的SNP對的集合,T代表集合S中SNP對的個(gè)數(shù)。
這樣,每個(gè)RNA-seq數(shù)據(jù)樣本均產(chǎn)生一個(gè)基于互信息的SNP信息值分布(I(si)(見圖4)。同樣,對這個(gè)樣本的每個(gè)SNV位點(diǎn)以同樣的方式(SNV對)求一個(gè)信息值,取95%的置信度,如果該位點(diǎn)的信息值落在SNP信息分布的置信區(qū)間外,則判定該位點(diǎn)是RNA編輯位點(diǎn),否則為SNP位點(diǎn)。
其次,為了提高RNA編輯位點(diǎn)判定的精確度,GIREMI用第一步中識(shí)別出的RNA編輯位點(diǎn)作為正樣本來訓(xùn)練廣義線性模型。該模型采用了兩個(gè)特征,一個(gè)是讀段中雜合SNP等位基因比率與SNV等位基因比率的差值的絕對值d,另一個(gè)則是基于序列本身特征的復(fù)合序列得分c(通過位置權(quán)重矩陣中+1和-1位置計(jì)算)。其回歸模型為:
(5)
其中,β0,β1,β2分別為需要學(xué)習(xí)的系數(shù),g為邏輯回歸函數(shù)。
該文只對GM12878數(shù)據(jù)集進(jìn)行了性能測試,并且達(dá)到了99.4%的準(zhǔn)確度,但是其準(zhǔn)確度的定義為100%-被預(yù)測為RNA編輯位點(diǎn)中SNP位點(diǎn)的百分比。
2.3 基于RNA-seq數(shù)據(jù)的有監(jiān)督學(xué)習(xí)方法
不同于GIREMI基于自主產(chǎn)生的正樣本來訓(xùn)練廣義線性模型,RED-ML和RDDpred是基于已知樣本進(jìn)行有監(jiān)督學(xué)習(xí)訓(xùn)練的RNA編輯位點(diǎn)檢測模型(見圖5)。

圖4 SNP與RNA編輯位點(diǎn)的信息分布[11]Fig.4 Information distribution of SNP and RNA editing sites[11]
圖5 RED-ML流程圖[12]
Fig.5 Flow chart of RED-ML[12]
Xiong等人[12]建立了一個(gè)基于機(jī)器學(xué)習(xí)的RNA編輯位點(diǎn)預(yù)測工具RED-ML,并選擇了邏輯回歸(Logistic regression)進(jìn)行模型的訓(xùn)練。RED-ML使用的特征有三大類。第一類是基本讀段特征,包括候選位點(diǎn)的支持讀段數(shù)量和計(jì)算出的編輯頻率。第二類特征與可能的測序失誤和錯(cuò)位有關(guān),包括支持讀段的圖譜質(zhì)量、候選位點(diǎn)在定位讀段中的相對位置、鏈偏差的指示、候選位點(diǎn)是否落入簡單重復(fù)區(qū)域等。第三類是基于RNA編輯的已知屬性,如編輯類型(是否為A-to-I),候選位點(diǎn)是否在Alu區(qū)域以及它的序列上下文。需要注意的是,與前兩類特征不同,第三類特征不能直接用來過濾非RNA編輯位點(diǎn)。然而,它們?nèi)匀豢梢蕴峁┯袃r(jià)值的信息,通過機(jī)器學(xué)習(xí)方法,將不同來源的信息結(jié)合起來做出分類決策。
作為一款RNA編輯檢測軟件工具,RED-ML的輸入是一個(gè)BAM文件,也可以利用相應(yīng)的基因組差異信息。RED-ML將提取候選RNA編輯位點(diǎn)及其相應(yīng)的特征,然后應(yīng)用邏輯回歸分類器以相應(yīng)的置信度檢測真正的RNA編輯位點(diǎn)。RED-ML可以僅基于人類RNA-seq數(shù)據(jù)執(zhí)行全基因組RNA編輯檢測,也可以利用匹配的DNA-Seq數(shù)據(jù),并與其他常見的RNA-seq數(shù)據(jù)分析步驟很好地結(jié)合。
該文用從其他文章中已發(fā)表的數(shù)據(jù)自己篩選出正負(fù)樣本進(jìn)行訓(xùn)練,其中隨機(jī)選取了80%作為訓(xùn)練集,20%作為測試集。在ROC曲線上的AUC達(dá)到了0.98,在PR曲線上的AUC達(dá)到了0.94。并且該文在CH24T、CH62T和HeLa樣本上做了RNA-seq驗(yàn)證實(shí)驗(yàn),取閾值為0.5時(shí)成功驗(yàn)證了90%的RNA編輯位點(diǎn)。
盡管RNA-seq數(shù)據(jù)可以用于RNA編輯位點(diǎn)檢測,但目前用RNA-seq進(jìn)行RNA編輯位點(diǎn)檢測的算法也具有相當(dāng)大的假陽性(False positive)風(fēng)險(xiǎn),這是用RNA-seq檢測RNA編輯位點(diǎn)的最大挑戰(zhàn)之一。由于短讀段誤對齊而產(chǎn)生的假陽性,本質(zhì)上是由以下幾種因素導(dǎo)致:(1)基因組序列固有的重復(fù)片段;(2)模糊的剪切連接;(3)個(gè)體之間普遍的多態(tài)性;(4)測序讀段的短缺。RDDpred(RNA/DNA Differences prediction)[14]是一種基于隨機(jī)森林算法的RNA編輯位點(diǎn)預(yù)測方法,能夠大大減少樣本中的假陽性數(shù)據(jù),從而提升RNA編輯位點(diǎn)的預(yù)測準(zhǔn)確率。
RDDpred首先對輸入的測序數(shù)據(jù)進(jìn)行初始比對,產(chǎn)生編輯位點(diǎn)候選者,然后從中選擇滿足特定條件的樣本作為訓(xùn)練數(shù)據(jù)(見圖6)。該方法使用RNA編輯數(shù)據(jù)庫RADAR和DARNED中的RNA編輯位點(diǎn)作為正樣本。而負(fù)樣本則通過MES(mapping error set)方法來收集,這種方法可以在比對時(shí)計(jì)算基因組內(nèi)的容易導(dǎo)致錯(cuò)誤的區(qū)域[13]。從RDD候選者中收集正/負(fù)樣本后,所有剩余的樣本被視為預(yù)測目標(biāo)。然后RDDpred建立了一個(gè)包含15個(gè)特征的隨機(jī)森林預(yù)測器來預(yù)測RNA編輯位點(diǎn)。

圖6 RDDpred 整體流程圖[14]Fig.6 Overall flow chart of RDDpred[14]
RDDpred用來自Bahn和Peng的小組進(jìn)行的獨(dú)立研究的兩個(gè)數(shù)據(jù)集進(jìn)行了評估[13, 15]。在Bahn的研究中,RNA-seq產(chǎn)生了115 132 348個(gè)讀段,RDDpred檢測到6 856 440個(gè)初始RDD并預(yù)測了105 564個(gè)RNA編輯位點(diǎn)。在Peng的研究,RNA序列產(chǎn)生了583 640 030個(gè)讀段,RDDpred檢測到58 666 976個(gè)初始RDD并預(yù)測3 076 908個(gè)RNA編輯位點(diǎn)。雖然這兩項(xiàng)研究都使用人體組織,但它們產(chǎn)生了不同數(shù)量的RNA編輯位點(diǎn)(105 564與3 076 908),這表明RNA編輯事件的表達(dá)模式在兩種環(huán)境下可能不同。而同時(shí),該方法用自己的模型驗(yàn)證了Bahn和Peng方法發(fā)現(xiàn)的編輯位點(diǎn),分別成功驗(yàn)證了95.32%(3 947/4 141)和90.37%(20 504/22 688),并且都大幅度減少了錯(cuò)誤的編輯位點(diǎn),NPV分別達(dá)到了84.21%和75.86%。
3 討 論
本文介紹了五種RNA編輯位點(diǎn)預(yù)測方法。第一種方法僅僅需要DNA序列。基于DNA序列的方法主要利用了序列間的相似性進(jìn)行預(yù)測,然而其缺點(diǎn)就是DNA序列中包含的信息是有限的,這使得其性能沒有達(dá)到較高的水準(zhǔn)(精確度大概在0.7左右)。此外,這種方法無法用于研究RNA編輯在不同條件(例如疾病)、不同個(gè)體、不同組織中的差異。
后四類方法則基于RNA-seq測序數(shù)據(jù)和機(jī)器學(xué)習(xí)模型,它們的共同特點(diǎn)就是高度依賴于高通量測序的質(zhì)量與深度。與此同時(shí),還有另外一個(gè)因素也限制了這些方法的效果,那就是RNA編輯水平,如果該位點(diǎn)處于一個(gè)比較低的編輯水平,那么預(yù)測難度將會(huì)大大提升。
RED-ML方法提出了大量可能與RNA編輯事件有關(guān)的特征,通過邏輯回歸模型進(jìn)行整合。它的一個(gè)缺陷就是模型對訓(xùn)練數(shù)據(jù)的依賴性很高,例如用人類數(shù)據(jù)得到的模型,在其他動(dòng)物上的預(yù)測效果并不理想。而且這種方法對于比對工具有很強(qiáng)的依賴性(目前版本只優(yōu)化了BWA和TopHat2),這導(dǎo)致用戶在選擇不同的比對軟件時(shí)會(huì)產(chǎn)生截然不同的結(jié)果阻礙研究的進(jìn)行。
如何準(zhǔn)確地區(qū)分SNP和RNA編輯,這是RNA編輯位點(diǎn)識(shí)別的一個(gè)核心問題。GIREMI方法通過計(jì)算不同SNV位點(diǎn)之間的互信息能夠更準(zhǔn)確地區(qū)分SNP和RNA編輯。但是其代價(jià)就是計(jì)算量的增加,如果考慮到時(shí)間成本的話,可能會(huì)對效率有所影響。并且要求對測序讀段的長度不能太短,否則無法覆蓋兩個(gè)感興趣位點(diǎn)。
RDDpred方法通過RNA編輯數(shù)據(jù)庫和MSE對數(shù)據(jù)進(jìn)行了篩選,然后用隨機(jī)森林模型進(jìn)行訓(xùn)練。該文章主要提出了組織特異性導(dǎo)致錯(cuò)誤正樣本的問題,因?yàn)樵赗ADAR和DARNED數(shù)據(jù)庫中有97%的編輯位點(diǎn)都是只存在于一個(gè)組織中,如果不清楚地將其篩選出來將會(huì)導(dǎo)致“預(yù)測危機(jī)”,因?yàn)檫@將從根本上(樣本上)導(dǎo)致訓(xùn)練失敗,從而降低預(yù)測性能。如果將其剔除假陽性的方法引用到其他模型中,或許會(huì)產(chǎn)生更好的效果,值得讓人期待。
4 總 結(jié)
RNA編輯事件的識(shí)別對于理解轉(zhuǎn)錄后調(diào)控是非常重要的。本文首先介紹了RNA編輯的概念和意義,然后介紹了兩個(gè)現(xiàn)有的RNA編輯數(shù)據(jù)庫(DARNED、RADAR)。而隨著機(jī)器學(xué)習(xí)的發(fā)展,其在RNA編輯的相關(guān)研究中也起到了重要的作用,故本文對已有的RNA編輯位點(diǎn)預(yù)測方法進(jìn)行了概述與討論,得到以下結(jié)論:(1)對于RNA編輯位點(diǎn),我們更關(guān)注其在樣本中的表達(dá);(2)對于RNA編輯來說,仍沒有一套像人類參考基因組一樣較為完備的標(biāo)準(zhǔn);(3)雖然已經(jīng)有了一些基于機(jī)器學(xué)習(xí)的預(yù)測RNA編輯位點(diǎn)的方法,但是并沒揭示RNA編輯的本質(zhì),即提取到判別RNA編輯位點(diǎn)的本質(zhì)特征。所以RNA編輯領(lǐng)域的研究還有很多亟待解決的問題和現(xiàn)象,希望以后能夠通過更深層次的模型去解釋RNA編輯,從而促進(jìn)相關(guān)疾病的研究以及精準(zhǔn)醫(yī)療的發(fā)展。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(bào)(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
