抗流感藥法匹拉韋雜質的合成
2019-04-12 05:32:02鄧玉曉段崇剛林治秘任業明孫晉瑞
食品與藥品 2019年2期
鄧玉曉,段崇剛,林治秘,李 丹,任業明,孫晉瑞
(山東省藥學科學院 山東省化學藥物重點實驗室,山東 濟南 250101)
法匹拉韋(favipiravir,商品名為Avigan),化學名為6-氟-3-羥基吡嗪-2-甲酰胺,是由日本富士膠片旗下富山化學工業公司研發的一種新型流感治療藥物,2011年3月在日本完成Ⅲ期臨床試驗,2014年3月被日本政府批準上市[1]。2014年3月,日本厚生勞動省批準其用于治療新型和復發型流感[2]; 2015年7月13日被臺灣批準上市用于治療流感;2016年6月14日被幾內亞批準用于治療埃博拉病毒[3]。本品為病毒RNA聚合酶抑制劑,能選擇性阻斷病毒RNA的合成,對哺乳動物細胞內的RNA合成不會產生任何抑制作用,因此,是一種安全有效的抗病毒藥物[4]。由于其特定的作用機制,除流感病毒外,法匹拉韋還可對抗其他多種RNA病毒,如HIV、黃熱病、SARS、埃博拉等[5]。
目前,文獻[6-8]報道的路線中,具有工業化前景的為3,6-二氯吡嗪-2-甲腈(化合物2)在相轉移催化劑四丁基溴化銨(Bu4N+Br-)的催化下,與無水氟化鉀發生雙氟取代反應,得3,6-二氟吡嗪-2-甲腈(化合物3);再與醋酸和三乙胺作用生成6-氟-3-羥基吡嗪-2-甲腈(化合物4);最后在濃硫酸作用下水解生成法匹拉韋。合成路線詳見圖1。
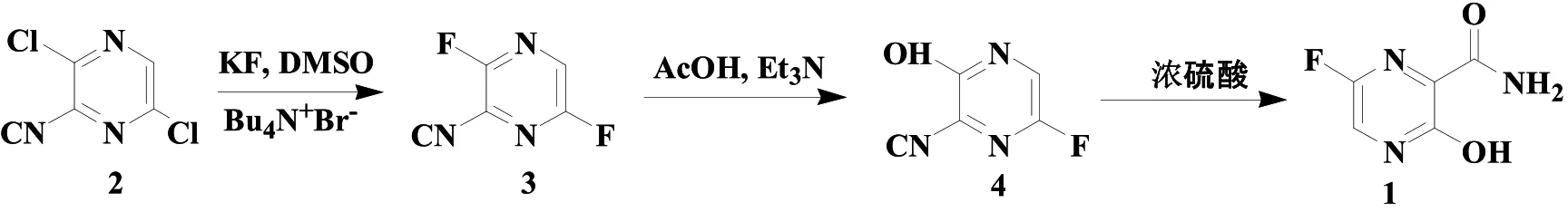
圖1 法匹拉韋的合成路線
筆者查閱了大量文獻,均未檢索到法匹拉韋雜質相關報道,為了控制法匹拉韋的質量,必須建立嚴格的質量標準,經大量的實驗探索和求證,最終確證了兩個雜質:6-氯-3-羥基吡嗪-2-甲酰胺(雜質1)和6-氟-3-羥基吡嗪-2-甲酸(雜質2),同時研究了相關雜質的制備方法,以為該產品的相關技術人員提供參考。
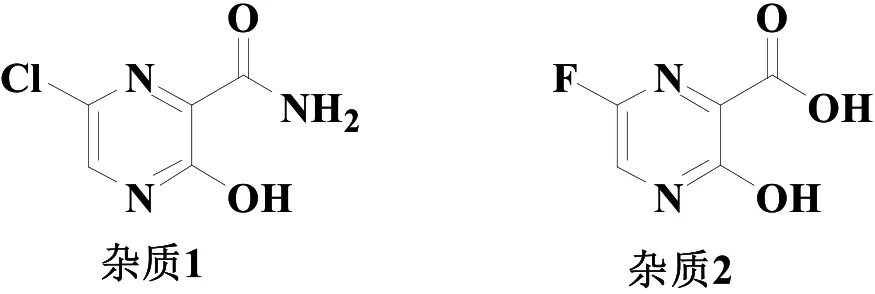
圖2 法匹拉韋合成中兩種主要雜質
從化學結構與分子活性角度分析,化合物2在3位和6位均有一個氯原子取代,然而,與兩個氯原子直接相連的碳原子的親電活性卻相差較大,3位碳原子由于受到芳香環上相鄰氮原子和鄰位氰基的雙重吸電子作用,電子云密度較低,極易受到氟離子的進攻,發生氟代反應; 6位碳原子受到芳香環上相鄰氮原子的吸電子作用,而氰基卻位于其間位,對其吸電子作用較小,電子云密度相對較高,所以,在相同條件下,有可能會產生3位氟代反應完全,而6位氟代反應不完全的情況,即生成3-氟-6-氯吡嗪-2-甲腈(化合物5),該化合物在冰醋酸和三乙胺作用下生成6-氯-3-羥基吡嗪-2-甲酰胺(化合物6),最后在濃硫酸作用下生成雜質1。由于化合物3中副產物5的含量較低,且在法匹拉韋原料藥合成過程中轉化為雜質1,并未在法匹拉韋原料藥中檢測到化合物5和化合物6。雜質1的來源流程圖詳見圖3。化合物4中2位氰基在酸性或堿性條件下水解,通過調節濃硫酸的濃度和反應溫度可控制反應主產物為法匹拉韋,但是過度水解產物雜質2卻很難避免,這一點在相關文獻[9]中已有詳盡報道。
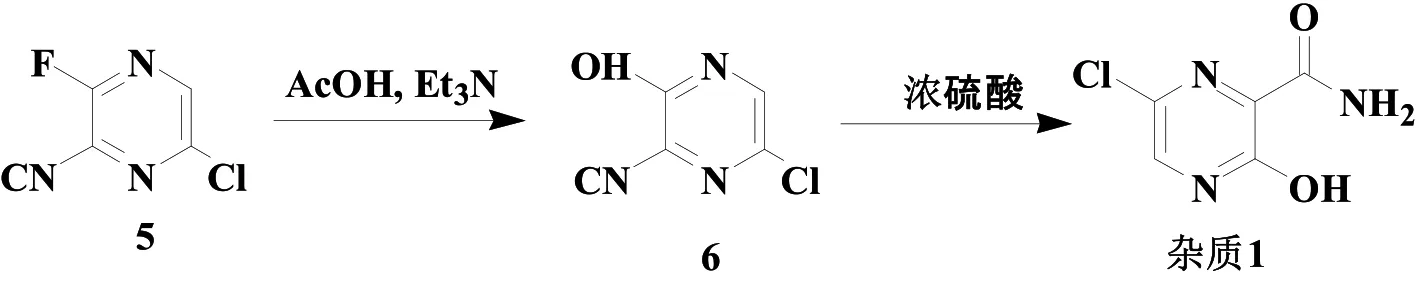
圖3 雜質1的來源流程圖
1 合成路線
1.1 雜質1的合成路線
相關文獻[10-11]報道了雜質1的合成方法,即以6-氯-3-羥基吡嗪-2-甲酸甲酯為起始原料,通過酯的氨解得雜質1。但酯的氨解反應速度慢且反應進行不徹底,所得產物中必然包含未反應完全的原料,且由于反應原料和產物極性相近,純化困難。合成路線詳見圖4。
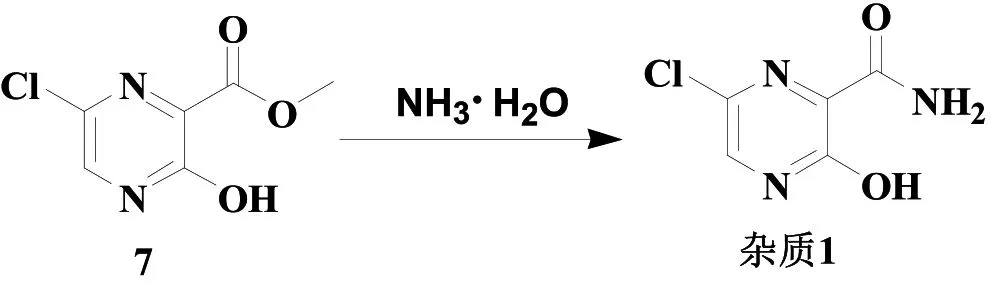
圖4 文獻[10-11]中雜質1的合成路線
本文采用3,6-二氯吡嗪-2-甲酰胺為起始原料,經親核取代反應、水解反應得到雜質1。起始原料易得,且反應條件溫和,產物易分離純化。詳見圖5。
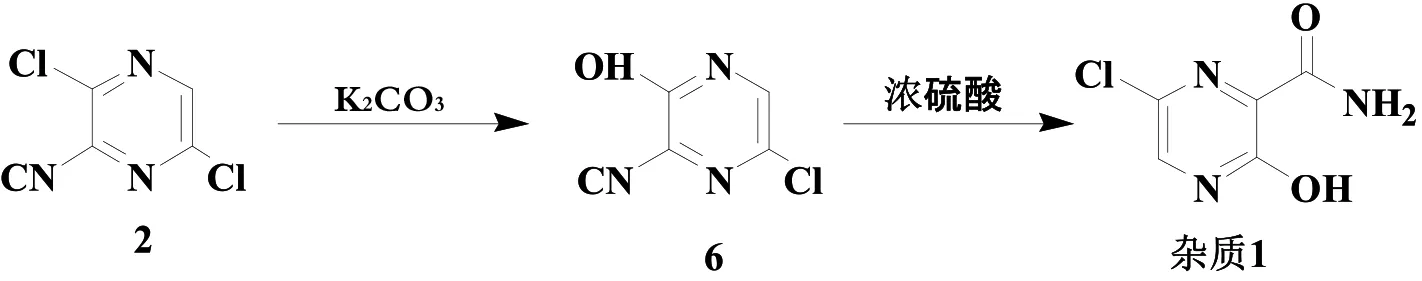
圖5 本文雜質1的合成路線
1.2 雜質2的合成路線
筆者查閱大量文獻,未發現雜質2的相關報道。我們以化合物4為起始原料,采用氫氧化鈉水溶液水解,然后用鹽酸調節反應液pH<3,即得雜質2。
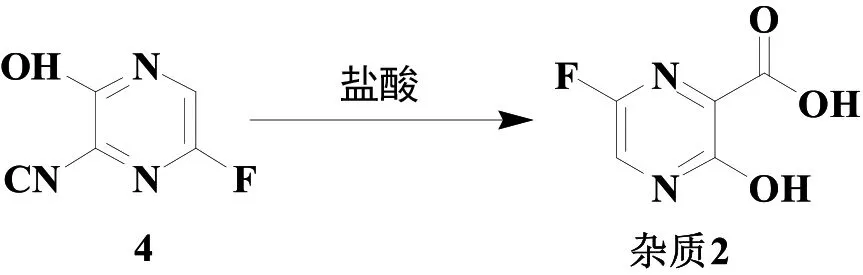
圖6 雜質2的合成路線
2 儀器與材料
2.1 儀器
Varian INOVA-300 核磁共振波譜儀(美國瓦里安公司);4000 Q TRAP 質譜儀(美國AB SCIEX公司);IKA HB 10 旋轉蒸發儀(德國IKA);循環水式多用真空泵(鄭州長城);DZF型真空干燥箱(北京市永光明醫療儀器廠);Agilent 1100高效液相色譜儀(美國安捷倫)。
2.2 藥品與試劑
3,6-二氯吡嗪-2-甲腈(四川南部縣誠信公司,純度>98.5%);6-氟-3-羥基吡嗪-2-甲腈(四川南部縣誠信公司,純度>98.5%);二氯甲烷(天津富宇,分析純);濃硫酸(國藥集團,分析純);無水碳酸鉀(國藥集團,分析純);N,N-二甲基甲酰胺(國藥集團,分析純);乙酸乙酯(天津科密歐,分析純);二甲苯(天津富宇,分析純);無水乙醇(萊陽經濟技術開發區精細化工,分析純);氫氧化鈉(國藥集團,分析純);無水硫酸鈉(國藥集團,分析純);濃鹽酸(國藥集團,分析純);石油醚(天津富宇,分析純,沸點60~90 ℃)。
3 方法與結果
3.1 6-氯-3-羥基吡嗪-2-甲腈(化合物6)的合成
取化合物2(5.0 g,0.029 mol)置入100 ml四口瓶,加無水碳酸鉀(7.94 g,0.057 mol),N,N-二甲基甲酰胺(25 ml),攪拌下油浴加熱,保持體系內溫度50~60 ℃,反應5 h。薄層色譜(TLC)顯示反應完全后,將反應液加入100 ml水中,用乙酸乙酯(100 ml×3)萃取,合并有機相,用水(100 ml×3)洗滌,經無水硫酸鈉干燥后過濾,濾液減壓蒸除溶劑,得黃色油狀物(4.13 g,收率:92.5%)。
3.2 6-氯-3-羥基吡嗪-2-甲酰胺(雜質1)的合成
向50 ml四口瓶中加入化合物6(4.13 g,0.027 mol),濃硫酸(10 ml),攪拌下油浴加熱,保持體系內溫度55~65 ℃,反應1 h。TLC顯示反應完全后,將反應液加入冰水(50 ml)中,用二氯甲烷(50 ml×3)萃取,合并有機相,用水(50 ml×3)洗滌,經無水硫酸鈉干燥后過濾,濾液減壓蒸除溶劑,得黃色固體(4.32 g,收率:93.6%),為雜質1粗品。向100 ml四口瓶中加入雜質1粗品(4.32 g),無水乙醇(80 ml),攪拌下油浴加熱,保持回流約0.5 h,關閉加熱和攪拌,自然降至室溫,抽濾,得淡黃色針狀結晶(3.70 g,收率:85.7%)。mp:200.5~201.6 ℃(文獻[12]mp:200~202 ℃),HPLC純度:99.6%。ESI-MSm/z:172.6[M-H]-。1H NMR(300 MHz DMSO-d6):8.61(s,2H),8.83(s,1H),13.66(s,1H);13C NMR(300 MHz DMSO-d6):169.5,157.9,142.5,140.3,128.4。
3.3 6-氟-3-羥基吡嗪-2-甲酸(雜質2)的合成
向100 ml四口瓶中加入化合物4(5 g,0.036 mol),4 mol/L鹽酸(20 ml),攪拌0.5 h,攪拌下油浴加熱,保持回流約3 h。TLC顯示反應完全后,將反應液加入冰水(50 ml)中,過濾,濾餅采用蒸餾水(50 ml×3)洗滌,60 ℃真空干燥至恒重,得棕色粉末(4.01 g,70.4%),為雜質2粗品。向100 ml四口瓶中加入雜質2粗品(4.01 g),石油醚(40 ml),室溫下打漿0.5~1 h,抽濾,得淡黃色粉末(3.87 g,收率:96.5%)。mp:178.8~180.2 ℃,HPLC純度:99.7%。ESI-MSm/z:157.0[M-H]-。1H NMR(300 MHz DMSO-d6):8.50(s,1H),13.7(s,1H);13C NMR(300 MHz DMSO-d6):172.5,161.2,142.3,138.2,127.0。[雜質1和雜質2的純度測定方法均為HPLC面積歸一化法,色譜條件:色譜柱:Intersil ODS-3,流動相:V緩沖溶液:V甲醇=45:55,緩沖溶液配置方法:0.5 g磷酸二氫鉀,1.0 g磷酸二氫鈉,加水1000 ml,搖勻即得,流速:1 ml/min,柱溫:30 ℃,進樣體積:20 μl,運行時間:40 min,檢測波長:238 nm]。
4 討論
4.1 本研究雜質1和雜質2的合成所采用的起始原料易得,反應條件溫和,未涉及高溫高壓超低溫等苛刻反應條件,反應的可操作性高。
4.2 采用重結晶或打漿的方法對2個雜質提純,收率高,操作簡便,避免了硅膠柱純化等繁瑣操作步驟。
4.3 本研究合成的2個雜質均純度高,性狀好,易于保存。
5 結論
本文分別以3,6-二氯吡嗪-2-甲腈和6-氟-3-羥基吡嗪-2-甲腈為起始原料,合成了法匹拉韋的兩個雜質,并通過MS、1H NMR、13C NMR等方法進行了結構確證。解決了2個雜質對照品來源問題,對法匹拉韋的質量控制有重要意義。
