同基因型同卵雙胞胎姐妹同患血卟啉病的不同臨床表現
2019-04-11 07:58:08王曉潔趙莘瑜焦淑潔王天舒劉曉宇
中風與神經疾病雜志 2019年3期
王曉潔, 趙莘瑜, 焦淑潔, 王天舒, 孫 琦, 劉曉宇
血卟啉病(Porphyria)是由于血紅素合成途徑中有關酶的缺乏導致卟啉類化合物代謝紊亂而發生的一種罕見疾病,以腹痛、神經精神癥狀及光感性皮膚損害為主要特征。血卟啉病高發于20~40歲女性,患病率(0.5~10)/10萬[1],臨床上以急性間歇性血卟啉病(acute intermittent porphyria,AIP)最為常見。本病患病率低,臨床表現各異,誤診率高,目前對該病的認識與研究仍較局限。查閱文獻尚無同卵雙胞胎同患血卟啉病的相關報道。現就我院收治的一對同卵雙胞胎姐妹同患AIP的相關臨床資料分析如下。
1 臨床資料
1.1 例1(姐姐) 女,24歲,以“腹痛14 d,抽搐4 d”于2017年12月9日入我院神經重癥病區。14 d前不明誘因出現發熱、腹痛,熱峰達37.8 ℃,停止排便,伴納差、進食少,偶伴惡心、嘔吐,嘔吐物為胃內容物。1 w前服用食用油排便后癥狀稍緩解。4 d前無誘因出現發作性意識喪失,四肢抽搐、頭后仰、雙眼上翻、牙關緊閉,伴小便失禁,持續4~5 min緩解,當天共發作3次,無頭痛、肢體無力、精神行為異常。就診于當地醫院,腦MRI發現雙側額頂顳葉異常信號,腹部CT考慮不全腸梗阻,給予禁食及其他治療(不詳),近1 d腹痛加重,就診于我院。查體:血壓175/125 mmHg,心率120次/min,律齊,各瓣膜聽診區未聞及雜音。腹部平坦,腹肌輕度緊張,右側腹部壓痛明顯,肝脾未觸及,肝區叩擊痛陽性,腸鳴音可聞及。神經系統查體無異常。
入院后完善相關檢查:血常規:中性粒細胞80.7%↑;淋巴細胞11.0%;中性粒細胞6.86×109/L↑;電解質:鉀3.17 mmol/L↓;鈉132 mmol/L↓。尿常規:比重1.005↓。肝功能:谷丙轉氨酶85 U/L↑;谷草轉氨酶88U/L↑。心肌酶:肌酸激酶1600.0 U/L↑;CK同工酶MB19.00 U/L↑;N端腦利鈉肽309.08 pg/ml↑。血脂:總膽固醇5.80 mmol/L↑。同型半胱氨酸37.35 μmol/L↑。抗甲狀腺過氧化物酶抗體174.70 IU/ml↑;抗甲狀腺球蛋白抗體264.00 IU/ml↑。腦脊液常規、生化、病毒、細菌、結核、寄生蟲、電泳檢查無異常。腎功能、結締組織全套無異常。性激素:睪酮1.00 ng/ml↑。皮質醇節律:皮質醇上午8點<1.00 μg/dl↓。胸部、全腹部增強CT:雙下肺慢性炎癥,未見腸梗阻。彩超:甲狀腺左側葉低回聲結節(TI-RADS分級:3級)甲狀腺左側葉囊實性結節;心臟、肝膽胰脾、雙腎、頸動脈、雙下肢深靜脈、腹主動脈彩超無異常。頭部MRI平掃+增強:雙側額頂顳枕葉皮質及皮質下見片狀腦回樣長T1長T2異常信號影,FLAIR呈高信號,DWI高b值呈稍高信號,雙側額頂顳枕葉異常信號未見明確強化。給予抗癲癇、營養神經、改善循環、抗感染、清除自由基、維持電解質平衡等對癥治療。
入院2 d查體四肢肌力4級,四肢腱反射消失。3 d四肢肌力3級,腱反射消失。患者癥狀進行加重,間斷腹痛、癲癇發作、自主神經功能紊亂、肌力減弱、腱反射消失,結合患者病史、癥狀、體征及相關檢查,考慮患者血卟啉病可能性大。入院5 d(家屬強烈要求轉至我科),精神差,反應遲鈍,計算力差,雙側軟腭上抬無力、咽反射減退,四肢肌張力低,雙上肢遠端肌力約3級,近端0級,雙下肢肌力1級。四肢腱反射未引出,病理征未引出,腦膜刺激征陰性。尿液實驗:尿液日光下暴曬后由洗肉水色變為深紅色(見圖1)。考慮血卟啉病,給予高糖飲食,靜脈滴注10%葡萄糖、營養神經、補充營養等對癥治療。入院12 d,雙上肢遠端肌力降至2級。加用激素治療,癥狀緩解仍不明顯。入院14 d出現呼吸表淺,三凹征陽性,咳嗽反射明顯減弱。雙上肢肌力1級,雙下肢踝關節上7 cm以下、雙上肢腕關節以上5 cm以下痛覺過敏。病變累及呼吸肌,有呼吸衰竭風險,再次轉入神經重癥必要時氣管切開。入院20 d復查頭部MRI:雙側頂葉見斑點狀等T1等T2信號,黑水像呈等信號,DWI呈高信號。病變呈現可逆性改變(見圖3~圖5)。患者家屬拒絕繼續治療出院。
1.2 例2(妹妹) 女,24歲,以“間斷腹痛5 y,再發并加重3 d”為主訴于2017年12月18日入院。5 y前進食后出現臍周疼痛,持續隱痛,可耐受,無惡心嘔吐,無發熱、腹瀉。后上述癥狀間斷發作,1 y前來我院查腹部平片:腸脹氣,以“腹痛原因待查”住消化科,查腹部CT:盲腸、升結腸、橫結腸明顯擴張積液、積氣。腹部彩超、腹主動脈、腸系膜動、靜脈無異常。頭部MRI:左側腦室三角區旁可見小片狀長T1長T2信號,黑水像呈高信號,DWI高b值未見彌散受限(見圖6)。給予禁食、灌腸、抗感染等治療后好轉出院。5 y來間斷腹痛,常為臍周持續隱痛,自服藥物效差,持續3 w可自行緩解。3 d前無誘因再次出現腹痛,較前加重。查體無異常。入院后查:血常規:白細胞5.80×109/L;紅細胞3.79×1012/L↓;血紅蛋白102.0 g/L↓;血小板112×109/L↓;紅細胞壓積0.302 L/L↓;平均紅細胞體積79.50 fl↓;平均紅細胞血紅蛋白含量26.80 pg↓。肝功能:前白蛋白102 mg/L↓。腎功能:尿素8.30 mmol/L↑;肌酐130 μmol/L↑;尿酸970 μmol/L↑。甲狀腺功能:T3:5.99 pmol/L;T4:33.83 pmol/L↑;TSH:0.01 μIU/ml↓。同型半胱氨酸39.70 μmol/L↑;葉酸、維生素B12、糖化血紅蛋白、電解質、傳染病檢查無異常。心電圖:竇性心動過速。尿液實驗:太陽暴曬下呈葡萄色(見圖2)。給予10%葡萄糖、降同型半胱氨酸等治療后,腹痛癥狀明顯緩解出院。
應用高通量測序方法,分析例1卟啉病相關致病基因HMBS、PPOX、HFE、ALAD、FECH、ALAS2、UROD、UROS、CPOX基因各外顯子編碼區及剪接區的變異情況;應用PCR結合Sanger測序技術分析例1、例2及患者父親HMBS基因c. 518G>A(p. Arg173Gln)位點的變異情況。結果:例1、例2及患者父親均存在HMBS基因c. 518G>A(p. Arg173Gln)雜合變異(見圖7)。
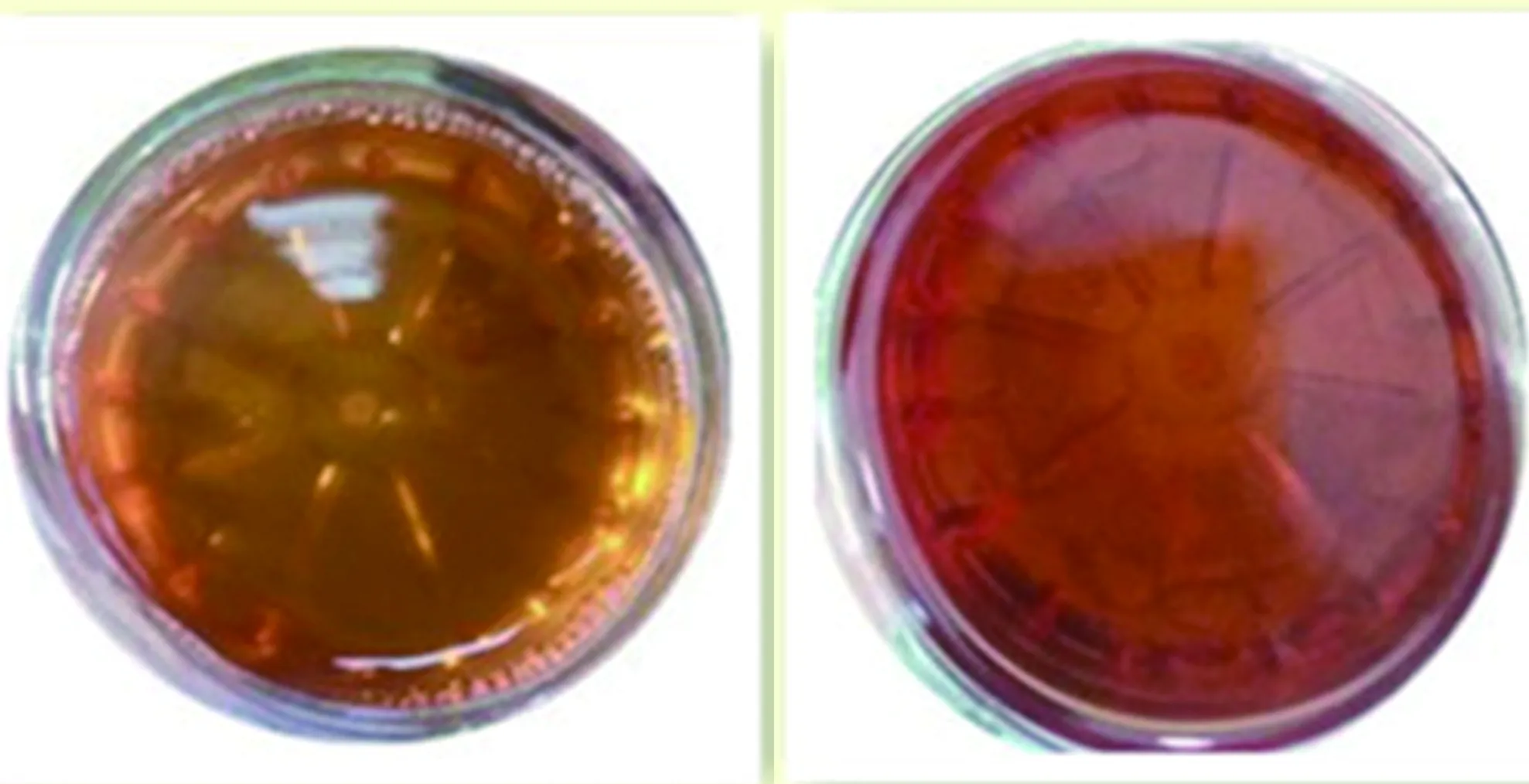
圖1 例1尿液陽光照曬前后對比(左圖為曬前,右圖為曬后)

圖2 例2尿液陽光照曬前后對比(左圖為曬前,右圖為曬后)

圖3 例1MRI T1、T2、Flair序列前后對比(第一排為治療前:廣泛額頂顳枕葉病變。第二排為治療后:頂葉斑點狀病變)

圖4 例1 MRI DWI序列前后對比(第一排為治療前:廣泛額頂顳枕葉病變。第二排為治療后:頂葉斑點狀病變)

圖5 例1第一次磁共振增強圖(廣泛額頂顳枕葉病變)
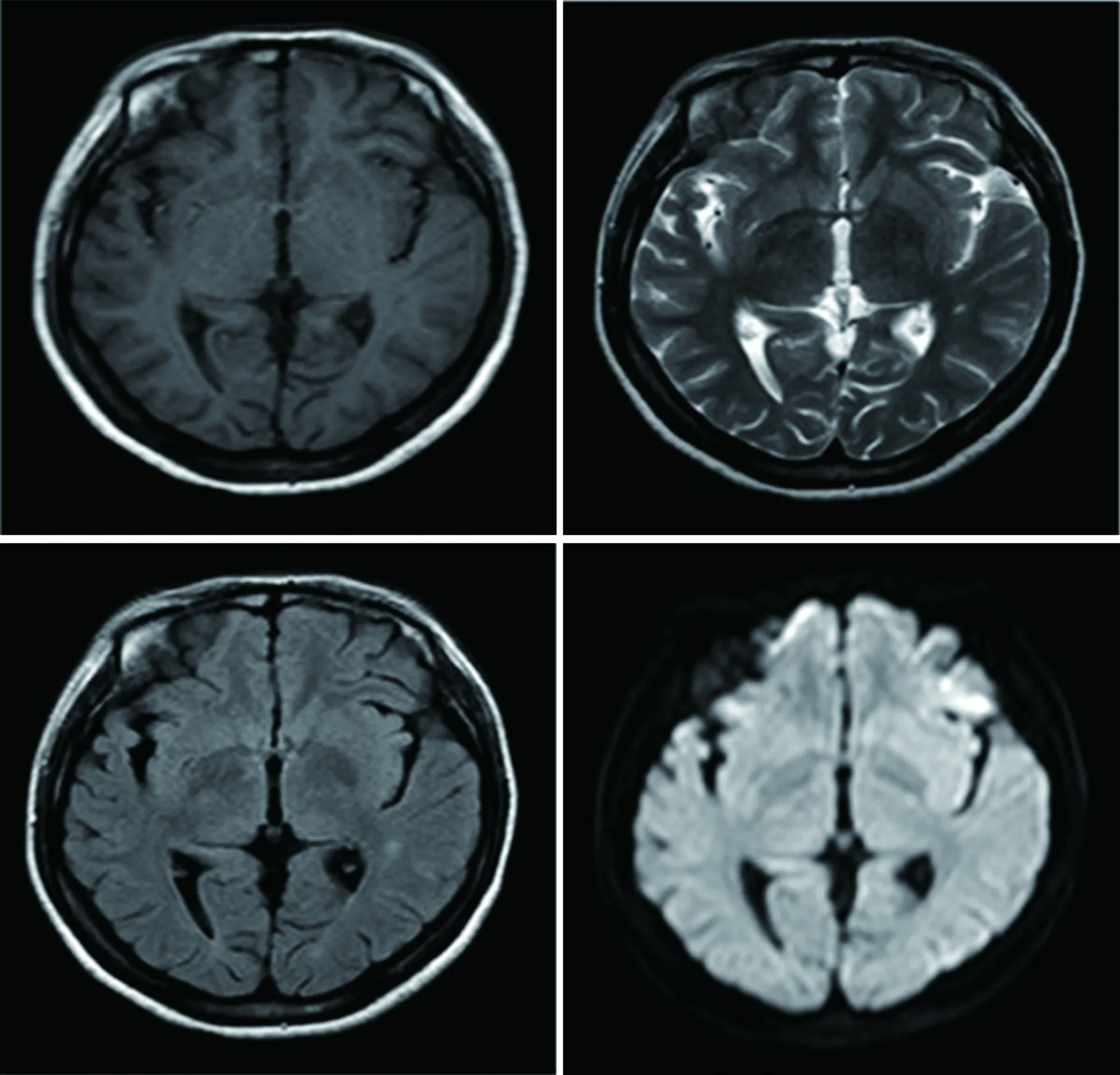
圖6 例2頭部MRI左側腦室三角區旁小片狀缺血灶

圖7 患者父親、例1、例2基因圖 (上圖為患者父親,中圖為例1,下圖為例2) 同為c. 518G>A(p. Arg173Gln)雜合變異
2 討 論
本組2例為雙胞胎姐妹,兩人長相、身材極為相似,據其家屬提供信息兩人為同卵雙胞胎。例1于24歲發病,以腹痛為首發癥狀,后迅速累及中樞神經系統、周圍神經系統并自主神經紊亂。查頭部MRI示雙側額頂顳枕葉皮質及皮質下異常信號影。給予對癥及高糖治療后,癥狀改善不明顯,逐漸累及呼吸肌,呼吸表淺、呼吸費力。20 d后復查頭部MRI:雙側頂葉點狀彌散受限。例2于19歲起出現間斷腹痛,病程5 y,未累及中樞、周圍神經系統,無精神癥狀。頭部MRI:左側腦室三角區異常信號,考慮缺血灶。給予高糖治療后,腹痛明顯緩解。二者尿液暴曬后均成酒紅色。因條件限制,二者均未應用血紅素治療。患者母親因病去世(不詳),其父女三人行DNA檢查,三者均為同一基因型。綜上所述,盡管同卵雙胞胎姐妹同患AIP,其臨床、影像學表現及預后完全不同。推測原因:(1)疾病嚴重程度除與基因型有關外,還受環境及其他因素影響。(2)發病累及中樞神經系統,影像學表現明顯者病情較重,盡管影像學表現可逆轉,但高糖等相關治療不能逆轉已有損害。
血卟啉病(Porphyria)是因血紅素合成路徑中有關酶的缺乏導致卟啉類化合物代謝紊亂而發生的疾病,依據血紅素前體物質(ALA、PBG 以及卟啉類化合物)異常合成或蓄積的主要組織部位,將卟啉病分為肝性血卟啉病和紅細胞生成性血卟啉病。肝性血卟啉病根據臨床表現的急劇程度又分急性和慢性兩類。其中急性肝性血卟啉病包括急性間歇性卟啉病(AIP)和遺傳性糞卟啉病(HCP)、混合型血卟啉病(VP)及ALA 脫水酶缺乏型血卟啉病(ALADP)。慢性肝性血卟啉病包括PCT和肝紅細胞生成性卟啉病(HEP)。紅細胞生成性卟啉病可分為先天性紅細胞生成性血卟啉病(CEP)和紅細胞生成性原卟啉病(EPP )。肝細胞線粒體的ALA合成酶(ALAS)催化甘氨酸和琥珀酸輔酶A 合成5-氨基乙酰丙酸(ALA),ALA從線粒體內轉入胞質中,兩個ALA 分 子 在 ALA脫 水 酶 作 用 下 合 成 卟 膽 原(PBG),四分子的PBG 在PBG脫氨酶作用下聚合生成線性結構的羥甲基膽色素(HMB),生理狀態下肝細胞內的酶催化HMB環化生成尿卟啉原Ⅲ,少部分生成尿卟啉原Ⅰ。尿卟啉原Ⅲ經脫羧酶作用生成糞卟啉原Ⅲ,后經糞卟啉原Ⅲ氧化酶作用轉化為原卟啉原Ⅲ,其在原卟啉原Ⅲ氧化酶作用下生成原卟啉原Ⅸ(PP),最后血紅素合成酶(即鐵螯合酶)催化亞鐵離子與PP合成血紅素。急性間歇性血卟啉病是最常見的一種類型,AIP 是由于血紅素生物合成途徑中PBG脫氨酶缺乏以至于血紅素前體物質ALA 及PBG的合成增多,蓄積在肝臟中,隨血液循環進入組織,尿中排泄增多。血紅素合成途徑中的第一個關鍵酶 5-氨基酮戊酸合成酶(ALAS-1)的誘導或血紅素合成需求量增大而肝臟 ALAS-1反饋抑制減弱可導致 ALA、PBG的蓄積合成明顯增多,導致臨床癥狀急性發作[2]。AIP 的年發病率為(2~3)/10萬,多見于成年女性,飲酒、感染、緊張、焦慮、月經來潮、苯巴比妥類藥物等均可誘發或加重 AIP[3]。不同誘發因素都是通過直接或間接激活血紅素合成過程中肝臟細胞內ALA 合成酶活性,引起卟啉及其前體物質在組織、循環中蓄積。女性患者發病機制尚不清楚,黃體酮、細胞色素酶P450、部分神經遞質都可引起肝臟異常代謝,誘發卟啉病急性發作,導致神經功能紊亂及肝臟損傷[4]。本文兩例均無誘因,例1性激素檢查睪酮升高,查閱相關文獻,尚無血卟啉病性激素異常的相關報道。
AIP臨床表現各異,以腹痛和神經精神癥狀為主[5],后者可累及中樞、外周、自主神經系統。(1)腹痛:是AIP患者最常見的一種癥狀,其特點是疼痛較重而彌散,呈進行性加重的腹部絞痛,可伴腹肌緊張、壓痛、反跳痛,伴或不伴膀胱區及后背部放射痛,中腹部較常見,持續時間數小時至數天不等。常伴惡心、嘔吐、腹肌緊張、便秘等,便秘可能與膀胱肌麻痹尿潴留有關。(2)中樞神經系統癥狀:意識障礙、癲癇、焦慮、抑郁、失眠、幻覺等。癲癇發病率高達5%,可能由低鈉血癥或不恰當用藥引起。(3)周圍神經系統癥狀:以運動癥狀為主,多首先累及雙上肢近端,表現為肌無力肌痛,后可漸累及四肢、延髓肌,致四肢癱瘓、呼吸無力,需呼吸機輔助通氣,嚴重者可致死亡。感覺癥狀較少見,表現為神經痛、遠端感覺缺失、近端肢體“穿泳衣”感。在急性發病期,腹痛常伴有一過性的上述肢體感覺癥狀。此外,腦神經病變多發生在并發神經系統癥狀的血卟啉患者,發生率達75%,且通常發生在肢體及軀干受累后,常累及面神經、迷走神經,三叉、舌下、副神經及動眼神經也可受累。視神經萎縮、眼肌麻痹、面神經癱瘓、吞咽困難、聲帶麻痹等也有報道。(4)自主神經癥狀[6]:包括心動過速、心律不齊、高血壓、尿潴留、低熱、出汗、脫水、電解質紊亂等。電解質紊亂多表現為低鈉血癥,發病率高達30%,推測原因,可能是由于下丘腦功能受損、抗利尿激素分泌胃腸道丟失過多引起[7]。經典的腹痛、神經病變、精神異常三聯征可發生于50%的急性發作患者中。單純出現神經精神癥狀而無腹痛癥狀者較少見,但的確存在單獨出現神經損害、腦病及精神癥狀的報道[8]。本文例1臨床表現較典型,腹痛、癲癇、易激惹典型AIP三聯征表現,四肢肌力減低、腱反射消失、痛覺過敏、呼吸無力等周圍神經癥狀,低血鈉、高血壓、竇性心動過速等自主神經紊亂。例2以腹痛為主要表現,并竇性心動過速,無精神神經系統表現。神經系統癥狀女性多見于男性,并且在青春期前及絕經后少見[9]。目前關于卟啉致神經病變主要有兩種假說:(1)卟啉類及其前體物質過度蓄積直接損害神經組織;(2)由于血紅素生物合成途徑中相關酶缺乏導致血紅素生成減少,細胞能量缺乏致軸突退化[10]。
血卟啉病的神經系統影像學表現及機制:主要有三種表現形式:(1)皮質與皮質下白質病變:兩側額葉、頂葉、枕葉皮質及皮質下白質(白質為主)斑片樣長 T1、長 T2信號影,DWI像上多呈低信號[11],ADC圖呈高信號,FLAIR像呈較高信號,病灶可融合。病變多發、多成對稱性改變,部分病變存在可逆性,與可逆性大腦后部白質腦病綜合征的病變分布和特點相似,二者可能存在相同的病理生理學機制[12]。也可能由于血卟啉病患者機體中血紅素缺乏導致NO產生減少(NO為一種重要的血管舒張因子)導致血壓升高和腦血管收縮[11]。(2)腦深部核團病變:雙側尾狀核頭及豆狀核、丘腦等深部灰質核團對稱性病變,病變呈長T1長 T2信號,FLAIR 像為高信號,DWI信號稍高,ADC像呈高或低信號[11]。可能與卟啉及其代謝前體物質對神經的毒性作用及頻繁癇性發作導致的缺氧性腦病有關。(3)腦缺血性病變:急性發作患者中,出現可逆與不可逆性缺血改變。可能與血管痙攣、血管收縮等有關。本文例1表現為皮質與皮質下白質病變,前后復查頭部磁共振,病變呈現可逆性改變。例2表現為點灶狀缺血性改變。
診斷及鑒別診斷:血卟啉病的診斷需綜合上述臨床、影像表現及相關輔助檢查[10]:AIP最重要的快速診斷方法是隨機尿PBG含量的定性及半定量測定。尿液檢測:ALA、PBG、尿卟啉明顯升高;血清、糞便:卟啉類物質輕度增高或正常。相關DNA基因突變位點檢測是診斷的金標準。AIP的遺傳方式是常染色體顯性遺傳,目前報道的有關AIP 基因突變位點有300多個,HMBS基因c. 518G>A(p. Arg173Gln)為已知致病性變異,但基因外顯率低,部分患者攜帶基因但不發病,基因型和表型無必然關聯,即某些突變并不代表疾病嚴重程度,疾病嚴重程度還受環境和其他因素影響[13]。即使攜帶同一致病基因的患者,其臨床表現也可有很大差別[14]。尿液實驗:尿液經加熱、加酸或暴曬后變紅甚至發黑。AIP患者肝臟 PBG 脫氨酶缺乏,多余的PBG經腎臟排出,PBG在尿液中可轉化成尿卟啉,可能導致尿液中尿卟啉、糞卟啉含量升高,最終卟啉類化合物及卟吩膽色素形成使尿液顏色加深。本文2例AIP患者及其父親驗證了上述觀點。雙胞胎患者及其父親均為同一基因型,符合AIP常染色體顯性遺傳,但三者臨床表現完全不同,例1病情重并進展迅速,例2病情較輕,高糖治療后癥狀迅速緩解,患者父親自述無明顯臨床癥狀,為AIP無癥狀攜帶者。追問患者父親,其兄弟姐妹及父母均無患者類似表現,因目前廣大民眾對遺傳病的諱莫如深,未能對其家系進行基因檢測。應做好相關宣傳教育,對發現血卟啉病患者的家系進行篩查,隨訪基因攜帶者,避免誘因,出現癥狀及時治療。最為重要的是對攜帶者進行產前診斷,優生優育,阻斷家系遺傳。
鑒別診斷:由于本病發病率低、較罕見且無特異性臨床表現,誤診率高,應做好鑒別診斷。(1)患者急性間歇性腹痛發作時,常被誤診為急性闌尾炎、急性胰腺炎、消化性潰瘍、膽結石、泌尿系結石等。急腹癥發作有各自特有的臨床特點,AIP腹痛發作時常伴有尿液實驗陽性[15]。(2)單純出現神經精神癥狀而無腹痛癥狀者較為少見,但確存在單獨出現神經損害、腦病及精神癥狀的報道,此時易與吉蘭巴雷綜合征、顱內病變等相混淆。AIP神經傳導研究表明以軸突損害為主,而吉蘭巴雷綜合征多為脫髓鞘病變[10]。顱內病變:顱內感染等可通過影像學及腦脊液檢查相鑒別,AIP患者腦部多見可逆性后部白質病變。
治療:(1)明確并避免誘因,如相關藥物的應用、飲酒、饑餓、感染等。(2)對癥治療:腹痛者可使用阿片類止痛劑;心動過速、高血壓可使用倍他洛克;惡心、嘔吐可使用奧氮平、勞拉西泮等,但多潘立酮、胃復安等應慎用;尿潴留者可導尿;癲癇發作可靜脈使用地西泮、左乙拉西坦,一般癲癇呈一過性發作,不需要維持治療[16];(3)血紅素療法:緩解癥狀快、效果好,常見不良反應有輕度凝血功能異常、血栓性靜脈炎、過敏性休克等。但目前我國大陸無血紅素制劑上市。(4)高濃度葡萄糖療法:高濃度葡萄糖(300 g~500 g)/d用于卟啉病輕度發作或是無條件使用血紅素時,可減少卟啉前體在組織中沉積,并可游離部分沉積于組織中的卟啉前體,但癥狀緩解不明顯者需要盡快使用血紅素治療,同時應積極防治低血鈉等電解質紊亂。(5)激素療法:孕激素、雌激素用于治療卟啉病療效不明顯,可誘發卟啉病,指南并不推薦使用。(6)肝移植療法:治療效果差、生活質量差、反復發作的患者,可考慮肝臟移植治療,嚴重的AIP患者肝臟移植成功后臨床癥狀明顯改善[17],但有相關移植風險。從本文2例的治療效果可見高糖治療對癥狀較輕者療效顯著,但對于重癥者療效欠佳。伴有神經癥候群的患者預后不良,應早發現,早治療,高糖治療效果不顯著時及時應用血紅素及其他療法。
AIP治療前景:重組人卟膽原脫氨酶(recombinant human-HMBS-enzyme,rh-HMBS) 是未來治療卟啉病的方法之一,rh-HMBS 以增加患者尿中卟啉物質排泄降低其在血中濃度而達到治療目的。如何使rh-HMBS 進入肝細胞并恢復肝細胞內 HMBS 的正常活性及血紅素的正常合成是當前階段研究重點[18]。RNA干擾治療在動物研究中證實有效,但臨床試驗效果不佳[19]。腺病毒介導的基因治療I期臨床試驗效果不顯著[20]。
通過報道上述兩例同基因型同卵雙胞胎姐妹同患血卟啉病的不同臨床表現,以期提高對血卟啉病認識。急性間歇性血卟啉病為常染色體顯性遺傳,相同基因型可有不同表型,預后亦有很大差別,應早發現,早治療,避免誘發因素。高糖治療對癥狀較輕者療效顯著,癥狀較重者需及時應用血紅素療法。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
中國生殖健康(2019年3期)2019-02-01 06:12:26
獸醫導刊(2016年6期)2016-05-17 03:50:35
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
