常染色體隱性遺傳念珠狀發一家系DSG4基因突變研究
2019-03-23 09:01:22任英豪陳晨曹瑞祥李昕張江安李小紅張貝于建斌孔祥東鄭州大學第一附屬醫院皮膚科45005鄭州大學第一附屬醫院遺傳與產前診斷中心45005
中華皮膚科雜志 2019年12期
關鍵詞:基因突變
任英豪 陳晨 曹瑞祥 李昕 張江安 李小紅 張貝 于建斌 孔祥東鄭州大學第一附屬醫院皮膚科 45005;鄭州大學第一附屬醫院遺傳與產前診斷中心45005
念珠狀發(monilethrix)是一種罕見的遺傳性皮膚病,表現為特征性毛干發育不良,即正常厚度的橢圓節間有規則的營養不良收縮,呈念珠狀外觀,薄的節間區域有很高的斷裂傾向。臨床表現為脫發伴毛囊角化過度和毛囊周圍紅斑,可僅累及枕部,嚴重時可累及整個頭皮、眉毛及睫毛。本病常染色體顯性遺傳(OMIM 158000),是由編碼毛發角蛋白的基因KRT81、KRT83、KRT86等雜合突變引起,而常染色體隱性遺傳是由橋粒芯糖蛋白4(DSG4)基因突變引起[1]。迄今為止,國內外[1-3]僅報道數例DSG4基因突變致常染色體隱性遺傳念珠狀發。本文報道1例DSG4基因復合雜合突變致常染色體隱性遺傳念珠狀發。
病歷資料
先證者女,3歲,漢族,出生后頭皮可見片狀毛囊角化性丘疹,無毛發生長。2歲左右開始生長稀疏毛發,粗細不均,間斷脫落,觸之易斷。毛囊角化性丘疹逐漸增多并擴展至雙眼瞼和枕部。發病以來,精神、飲食、睡眠和大小便正常。患兒系第2胎第2產,足月順產,無產傷窒息史,母乳喂養,預防免疫接種隨當地進行。患兒有1哥哥,表型正常,父母非近親結婚,毛發密度和分布正常。母親懷孕期間無服藥、接觸有害物質及病毒感染史。家族其他成員無類似疾病及其他先天性或遺傳病史。本研究經鄭州大學第一附屬醫院科研項目倫理審查委員會批準(2018-YB-05)。
體檢:營養良好,身高、體重及智力正常,其他系統檢查無明顯異常。皮膚科檢查:頭皮頂部、枕部片狀頭發稀疏、脫落,殘留斷發及毛囊角化性丘疹。病發及毛囊角化性丘疹在頭皮頂部及枕部明顯,部分眉毛、睫毛脫落、折斷,長短不一(圖1A~1C)。牙齒、指甲、趾甲及汗腺正常。
實驗室檢查
血常規示白細胞7.6×109/L,紅細胞4.39×1012/L,血紅蛋白122g/L,血小板411×109/L。血清IgA 0.41 g/L(參考值0.8~5 g/L),血清IgE 126.50 IU/ml(參考值0~60 IU/ml)。尿糞常規、肝腎功能、電解質、空腹血糖、凝血功能、紅細胞沉降率、降鈣素原、補體C3及C4、25羥基維生素D3、微量元素檢測均正常。乙型肝炎病毒、丙型肝炎病毒、梅毒、HIV篩查均未見異常。
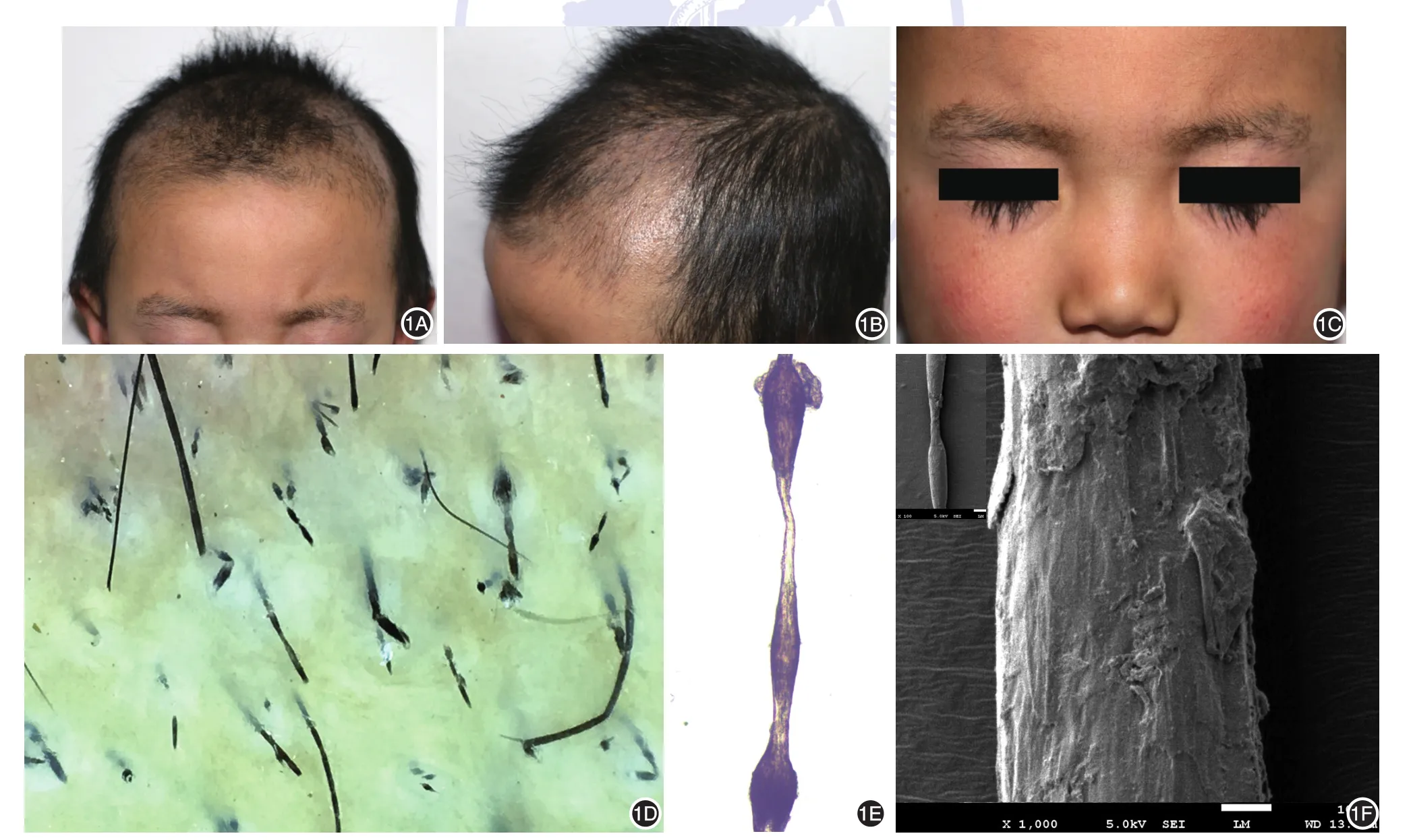
圖1 念珠狀發患兒臨床表現及皮膚鏡、光鏡和電鏡檢查結果 1A~1C:患兒頭皮頂部、顳部片狀頭發稀疏、脫落,殘留斷發及毛囊角化性丘疹,部分眉毛脫落、折斷,長短不一;1D:皮膚鏡下頭發毛干粗細不均,長短不一,可見黑點征樣改變,頭發毛干可見念珠狀外觀,結節間距較短(×10);1E:光鏡下見典型念珠狀發(×40);1F:掃描電鏡示典型念珠狀發(左上角小圖,×100),鱗片狀毛小皮脫落,毛皮質呈枯樹皮樣外觀(×1 000)
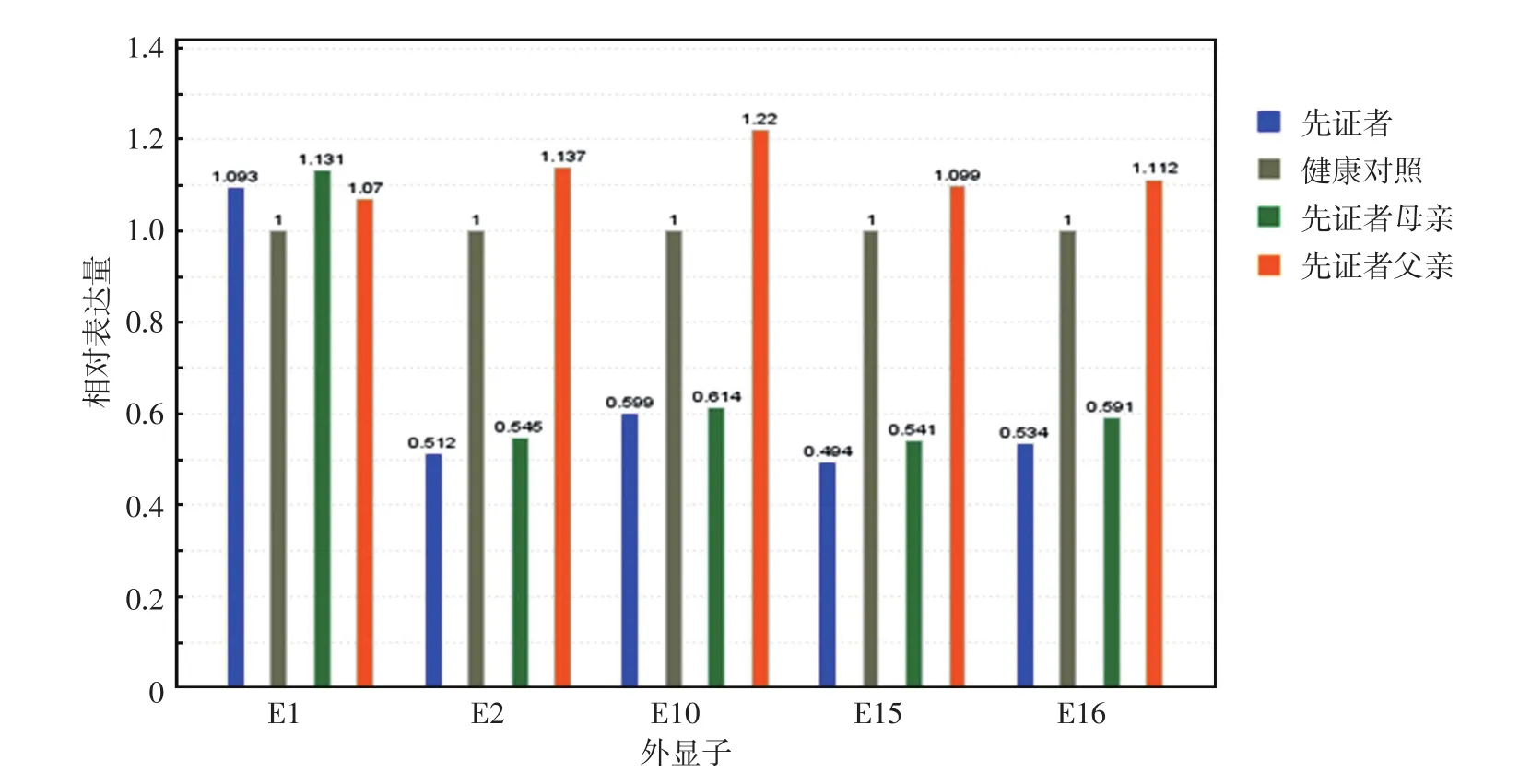
圖2 DSG4基因第1、2、10、15、16號外顯子實時熒光定量PCR檢測結果 先證者和其母親均第1外顯子正常,第2、10、15、16外顯子雜合缺失;先證者父親和健康對照第1、2、10、15、16外顯子均正常
皮膚鏡及毛發掃描電鏡檢查
皮膚鏡下頭發毛干粗細不均,長短不一,可見斷發殘根,呈黑點征樣改變,頭發毛干可見念珠狀外觀,結節間距較短,毛發很短,易于折斷(圖1D)。光鏡下病發呈典型的念珠狀外觀(圖1E)。征得患兒父母知情同意后,取患兒少許毛發,掃描電鏡(日本電子株式會社)觀察,結果顯示:毛發粗細不均,低倍可見典型念珠狀發,高倍示病變處鱗片狀毛小皮脫落,遺留毛皮質,呈枯樹皮樣外觀(圖1F)。
基因檢測
征得患兒父母知情同意后采集患兒及其父母血樣進行基因測序,以100例健康人外周血作為對照。采用北京邁基諾基因科技有限公司設計的皮膚病相關基因檢測包,對先證者皮膚病相關基因外顯子區及相鄰側翼序列進行靶向性捕獲,使用Illumina Nextseq500測序儀進行高通量測序,平均測序深度為300×,覆蓋度為95%。高通量測序結果顯示,先證者DSG4基因存在第2~16外顯子雜合缺失,以及第6外顯子c.574T>C(p.S192p)(NCBI序列號:NM-177986)突變,分別通過實時熒光定量PCR及Sanger測序得到驗證。先證者母親攜帶DSG4基因2~16號外顯子雜合缺失突變,先證者父親攜帶DSG4基因c.574T>C(p.S192p)雜合突變。100例健康人均未發現該位點變異。見圖2、3。
討 論

圖3 先證者及其父母和健康對照DSG4基因第6外顯子574位點正向測序結果 3A:健康對照為T/T純合峰;3B:先證者表現為c.574T>C(p.S192p)半合子變異:3C:先證者父親c.574T>C(p.S192p)雜合變異;3D:先證者母親為正常序列單倍體
橋粒屬于鈣黏蛋白超家族,由橋粒芯蛋白、橋粒膠蛋白和橋粒斑珠蛋白組成[4]。2003年Whittock和Bower[5]鑒定了一種新的人類DSG,將其命名為DSG4。Kljuic等[6]報道2例局限性常染色體隱性遺傳稀毛癥(localized autosomal recessive hypotrichosis,LAH),患者臨床表現與念珠狀發相似,DSG4基因存在5~8號外顯子缺失,為致病性突變。2006 年 Shimomura 等[7]、Zlotogorski等[8]和Schaffer等[9]分別報道伴有念珠狀發改變的LAH,患者臨床表型與念珠狀發相似,均發現DSG4基因突變,這表明DSG4基因突變引起的LAH在臨床上可以與念珠狀發重疊。2009年Bazzi等[10]發現DSG4基因突變導致人毛發減少,體外和體內實驗均證明DSG4定位于橋粒,在人類毛囊中具有高度特異的表達模式,對毛干完整性非常重要。
2011年Farooq等[1]報道1例DSG4基因突變致常染色體隱性遺傳念珠狀發,并發現突變的DSG4對橋粒斑珠蛋白親和力降低,首次展示了DSG4和橋粒斑珠蛋白的相互作用。2015年Wang等[11]報告1例DSG4基因突變致LAH,患者表現為稀疏毛發及毛囊角化性丘疹,眉毛和睫毛也受損,癥狀類似念珠狀發。王沛等[2]報告1例DSG4基因突變致常染色體隱性遺傳念珠狀發,患者DSG4基因突變從而形成翻譯提前終止密碼子,引起DSG4基因翻譯提前終止,該突變導致橋粒芯糖蛋白C端結構缺失,影響DSG和橋粒斑珠蛋白的連接,導致細胞間連接系統缺陷,從而出現念珠狀發,并指出LAH患者的DSG4基因突變報道都為純合突變,而念珠狀發患者幾乎均為雜合突變,與我們本次報道DSG4基因雜合突變導致念珠狀發一致。王沛等[2]和Wang等[11]均認為DSG4基因突變導致的LAH和念珠狀發可能為同一種疾病,但還需更多病例報告和突變研究來證實。本文先證者攜帶DSG4基因c.574T>C(p.S192p)(NM-177986)突變,與既往文獻[7]報道相同,該位點在人類DSG中完全保守,該突變為致病突變。同時,本文先證者DSG4基因2~16號外顯子缺失,檢索國內外文獻未發現有相同缺失突變的案例報道。既往文獻[6,12]報道LAH患者DSG4基因5 ~8號外顯子缺失,產生類似念珠狀發表現。考慮到患者DSG4基因存在大片段缺失突變,推測為致病性變異。綜上推測本例患兒攜帶的DSG4基因點突變和缺失突變產生異常的DSG4蛋白,導致細胞間連接系統缺陷,影響相鄰細胞間黏附,從而引起毛發角蛋白表達異常,患兒出現念珠狀發表型。
2016年Muhammad等[12]對6個LAH直系血親家族的DSG4基因序列分析發現,6個家族的受影響個體中都發生了5~8號外顯子缺失突變。這些家族的其他13種突變中,2種突變引起念珠狀發,3種導致念珠狀發和LAH并存,剩下的8種導致LAH,推測DSG4基因突變的表型變異可能與不同修飾基因的作用有關。
常染色體隱性遺傳念珠狀發和LAH的致病基因均為DSG4,臨床表現相似,表現為脫發、毛發稀疏等,差異為念珠狀發患者更容易出現角化過度的毛囊丘疹和毛囊周圍紅斑,毛干呈典型念珠狀發樣外觀。依據典型的臨床特征、家族史、實驗室檢查、光鏡、皮膚鏡、掃描電鏡檢查和基因檢測,本例患兒常染色體隱性遺傳念珠狀發診斷明確。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
