基于溫帶和熱帶玉米群體全基因組FST和XP-EHH的 選擇信號檢測
2019-03-18 06:07:38楊宇昕鄒棖
中國農業科學 2019年4期
關鍵詞:信號
楊宇昕,鄒棖
?
基于溫帶和熱帶玉米群體全基因組F和XP-EHH的 選擇信號檢測
楊宇昕,鄒棖
(中國農業科學院作物科學研究所,北京 100081)
【目的】玉米起源于熱帶地區,經過自然和人工選擇,廣泛的種植于溫帶地區。開花是玉米生長發育的中心環節,也是熱帶玉米向溫帶環境種植的主要適應性性狀。鑒定玉米在馴化過程中出現的受選擇基因區段,并進一步挖掘開花候選基因,為玉米的群體改良、開花遺傳機理解析提供數據支撐。【方法】首先單獨分析30份溫帶玉米自交系和21份熱帶玉米自交系的單倍型數據,通過過濾高缺失和等位基因頻率較低的變異位點,得到高質量的SNP(single nucleotide polymorphism)標記,利用SnpEff軟件對溫帶和熱帶玉米群體的基因組多態性位點進行了功能預測。其次過濾得到同時存在于溫帶和熱帶玉米的高質量SNP標記,對溫帶和熱帶玉米的基因型數據進行主成分分析(principle component analysis,PCA)以確定其群體結構,之后利用群體分化指數(fixation index,F)和群體間擴展單倍型純合度(cross population extended haplotype homozygosity,XP-EHH)法分析溫帶和熱帶玉米群體間的選擇信號分布情況,選擇F和XP-EHH值的top 1%為閾值,篩選得到受選擇位點。通過對SNP進行功能注釋得到溫熱帶玉米群體受到選擇的基因。利用agriGO工具對候選馴化基因進行功能富集分析。利用相關的生物信息學數據庫對候選基因進行功能注釋,進一步鑒定玉米馴化過程中的開花候選基因。【結果】通過對溫熱帶玉米群體的高測序深度的SNP進行分析,發現熱帶玉米群體的SNP數目為14 123 408個,溫帶玉米群體的SNP數目為8 791 673個,鑒定到的SNP主要分布于基因間區。2個群體中均存在的SNP標記數目是204 752個。主成分分析表明溫帶和熱帶玉米可以顯著的分為兩個類群。F選擇信號的top 1%是0.3593,共鑒定到557個候選馴化基因,XP-EHH選擇信號法的top 1%是3.2681,共鑒定到1 913個候選基因。鑒定到多個候選基因與玉米的開花調控密切相關,包括、、如抑制開花基因的表達,導致玉米在長日照環境下出現晚花表型,是一個重要的開花調控基因;COL1與開花促進因子FT蛋白互作,加速玉米開花以適應長日照環境;的功能注釋揭示該基因是一個光敏色素互作因子,與光周期基因互作。【結論】熱帶玉米群體具有更高的遺傳多態性,篩選到一系列參與了熱帶玉米和溫帶玉米的分化候選基因,并且重點挖掘了參與其中的玉米開花調控相關基因。
玉米;選擇信號;群體分化指數;群體間擴展單倍型純合度;開花基因
0 引言
【研究意義】玉米是非常重要的糧食和飼料作物,考古學和遺傳學研究證明現代玉米起源于約9 000年前的墨西哥西南部,其野生祖先是大芻草[1-3]。玉米的起源中心位于低緯度短日照的熱帶環境,經過不斷地人工選擇和馴化,逐漸降低了光周期敏感性,并且被廣泛種植于58°N到40°S的溫帶長日照地區[4-5]。根據玉米對地理環境的適應性,其可以劃分為適應長日照環境的溫帶玉米和適應短日照環境的熱帶玉米2種類型[6]。熱帶玉米不僅具有優良的農藝性狀(例如根系發達、抗病蟲性強[7]等特點),而且具有豐富的遺傳變異[8],是進行群體改良的優良材料。不同的玉米群體在人工選擇和馴化的過程中,逐漸適應當地的生長環境,導致染色體區段上控制目標性狀基因的優勢等位基因頻率逐漸增加,多態性發生改變,根據中性遺傳假說[9],這將會導致與目的基因緊密連鎖的染色體序列多態性也會隨之發生改變,這一個過程稱之為搭車效應[10]。由于人工選擇而導致的DNA結構變化稱之為即選擇信號(selection signal),其特點是染色體區段上出現較長距離的單倍型純合、連鎖不平衡值改變以及優勢等位基因頻率的增加。因此,利用選擇信號法探究溫熱帶玉米群體的基因組變化,可以鑒定在熱帶玉米適應性改良過程中受到選擇的基因區段,并且深入的挖掘開花基因,對于揭示玉米的馴化改良的遺傳機制具有重要意義。【前人研究進展】選擇信號檢測方法根據其分析原理可以分為以下3種類型:(1)基于等位基因頻率譜的方法,代表的計算方法包括Tajima's D[11]和CLR(composite likelihood ratio)[12]等;(2)基于連鎖不平衡的方法,主要包括EHH(extended haplotype homozygosity)[13]、iHS(integrated haplotype score)[14]和XP-EHH[15]等;(3)基于群體分化進行群體遺傳分化的方法,主要包括F法[16-18]。目前,應用較為廣泛的選擇信號檢測方法包括群體分化指數法(F)和群體間擴展單倍型純合度法(XP-EHH)。F統計理論最早由WRIGHT[16]提出,經過對其分析方法和計算理論的不斷改進,如今使用較多的是由WEIR等[17]提出的無偏估計的F。F選擇信號法可以掃描全基因組范圍內的SNP位點,計算每個SNP的F值,其取值范圍為0—1,0代表群體間所有位點都沒有出現分化,1代表群體間已經完全分化。XP-EHH選擇信號是基于基因的連鎖不平衡原理,群體經過人工選擇和改良,將會出現較大范圍的染色體重組,由于連鎖作用的存在,導致突變基因附近的中性位點也會逐代的傳遞,因此,在染色體上形成較長范圍的單倍型純合。XP-EHH的計算方法是EHH和iHS統計原理的擴展,相比于F方法只能計算出發生分化的位點,XP-EHH統計量可以得到選擇作用所在的群體,即XP-EHH統計值為正數時,表明選擇發生在試驗群體,反之則發生在參考群體。隨著越來越多物種的參考基因組構建完成,利用F和XP-EHH選擇信號法,已經在遺傳進化、基因挖掘等生物學領域取得了很大的進展。AXELSSON等[19]對狼和狗采取全基因組重測序得到3.8億個SNP遺傳變異標記,通過進行F選擇信號檢測,在淀粉消化和脂肪代謝中起到重要作用的10個調控基因表現出明顯的選擇信號,這些調控基因可以促進狗的祖先在以淀粉為主要食用能量的人類社會中生存,相比于肉食性習性動物的狼,可以得到更多食物,這種可以消化淀粉的適應性演化是狼馴化為狗的關鍵步驟。LIU等[20]利用溫帶、熱帶和亞熱帶玉米中代表性的260個玉米自交系,結合F的計算方法,發現熱帶玉米相比于溫帶玉米具有更高的遺傳多樣性和更多的等位基因位點。通過研究地方品種和自交系的位點間差異,表明現代玉米低于80%的位點來源于地方品種,為利用玉米地方品種進行改良提供了有力的試驗支持。HE等[21]利用XP-EHH選擇信號的方法,在溫帶玉米改良進程中一共鑒定到超過1 100個候選基因區段,通過基因富集分析,發現這些基因主要參與蔗糖合成和油分含量調控等。【本研究切入點】隨著高通量測序技術在農業領域的廣泛應用,高質量的玉米參考基因組圖譜[22-23]和單倍型圖譜[24-25]相繼構建完成,極大地促進了玉米功能基因組學的發展,同時這些公共數據也為廣大科研人員進行其他的生物學問題研究提供了寶貴的數據資源。熱帶玉米在經歷人工馴化和改良過程中逐漸適應溫帶環境,因此,有必要利用選擇信號法揭示在改良過程中發生選擇的位點,挖掘候選馴化基因,進一步從基因層面上探究玉米的群體改良。【擬解決的關鍵問題】本研究選擇具有代表性的30個溫帶玉米自交系和21個熱帶玉米自交系作為材料,基于全基因組單倍型數據,分析溫熱帶玉米基因組多態性,并且結合F和XP-EHH 2種方法進行群體間選擇信號的檢測,挖掘發生選擇的馴化基因,為玉米馴化的遺傳機理解析提供理論依據。
1 材料與方法
1.1 溫、熱帶玉米自交系基因型的獲取
選取具有代表性的30個溫帶玉米自交系和21個熱帶玉米自交系,其基因型數據來自于玉米單倍型圖譜第三版[25],選擇單倍型圖譜中測序深度較高的重測序數據,以確保其準確率。原始基因型數據以VCF文件存儲。考慮到原始基因型數據等位基因頻率偏低可能影響后續分析[26],因此,利用VCFtools軟件將缺失率較高和最小等位基因頻率低于0.05的SNP剔除,相關參數為--maf 0.05。首先進行溫帶玉米群體和熱帶玉米群體的SNP篩選,分別得到溫帶和熱帶的基因型數據,這兩個基因型數據只進行SNP的功能注釋。使用的選擇信號計算方法要求溫帶和熱帶玉米群體具有相同的SNP數目,因此,將溫帶玉米和熱帶玉米一起進行SNP的篩選,得到共存于溫帶和熱帶玉米群體的高質量SNP標記,該基因型用于F和XP-EHH選擇信號的計算。為了評估溫熱帶玉米群體全基因組水平上的變異情況,利用SnpEff[27](版本號4.3p)軟件對溫熱帶玉米群體的變異信息進行功能注釋。SNP密度分布利用R軟件包CMplot繪制。玉米參考基因組下載于Ensemble數據庫。
1.2 主成分分析
主成分分析選擇Tassel(版本號5)軟件。首先將過濾得到的溫熱帶玉米群體的基因型文件導入到Tassel軟件,按照5個成分進行分析,選擇前2個主成分進行繪圖展示。
1.3 FST選擇信號檢測
采用群體分化指數(F)法進行溫熱帶玉米間選擇信號的檢測,其計算原理是依據染色體等位基因頻率變化。群體間選擇信號的檢測可以揭示不同群體馴化過程中經歷的自然選擇。按照WEIR等[17]的統計方法進行F值的計算,考慮到基于單位點SNP掃描的方法容易受到遺傳漂變等因素的影響,因此,為降低假陽性,選擇滑動窗口的計算方法來增加選擇信號的靈敏度[28]。利用VCFtools軟件計算滑動窗口內的F值,相關參數設置為--fst-window-size 200 kb、--fst-window-step 100 kb。利用R包qqman展示全基因組水平上的F值。為了鑒定F值的受選擇位點,選擇F值的top 1%作為顯著閾值線[29],高于閾值線的SNP位點定義為“受選擇位點”,F的計算公式參考WEIR等[17]報道。
1.4 XP-EHH選擇信號計算
同時利用基于群體間擴展單倍型純合度(XP- EHH)的方法檢測群體間存在分化的基因區段。XP-EHH選擇信號的計算使用selscan[30]軟件(版本號v1.1.0),將溫帶玉米群體作為試驗群體,熱帶玉米群體作為參考群體。當基因組某一區段的XP-EHH的值為正,代表在試驗群體中發生了選擇,反之則表示參考群體的基因組片段發生選擇。基于XP-EHH選擇信號得到的統計值近似符合正態分布[15],對XP-EHH的值進行標準化正態分布處理,使用selscan的norm參數對原始的XP-EHH值進行標準化處理。將標準正態化處理后的XP-EHH值從大到小排序,取其top 1%[29]作為顯著的閾值線用以判斷溫、熱帶群體間是否發生選擇,并且篩選出受選擇基因區段,XP-EHH的計算原理參考SABETI等[15]的報道。
1.5 候選基因的鑒定
F法是按照滑動窗口計算得到的受選擇位點,因此,將滑動窗口的起始和終止位點各向上游和下游擴增50 kb作為受選擇選擇區段,使用Bedtools[31](版本號v2.26)軟件將受選擇位點附近的候選馴化基因與玉米參考基因組進行基因比對,編碼基因若是落在滑動窗口內則定義為候選基因。針對XP-EHH法得到的受選擇位點,以顯著SNP位點向上下游各擴增50 kb作為受選擇的區域,同樣利用Bedtools軟件篩選出候選基因。由于篇幅所限,利用maizeGDB(https://www. maizegdb.org/)對選擇信號值較高的基因進行基因功能注釋。
1.6 候選基因的富集分析
利用在線工具agriGO進行候選基因的富集分析[32]。分析方法采用奇異富集分析(singular enrichment analysis,SEA),參考數據庫選擇ssp. v5a,顯著性GO條目的檢驗使用Fisher精確校驗,顯著性閾值為0.05。分析內容包括細胞組分、生物過程和分子功能等。
2 結果
2.1 溫、熱帶玉米群體的SNP功能注釋
利用SnpEff軟件評估溫、熱帶玉米群體在基因組水平上的多態性情況(圖1),基因組注釋信息來源于B73自交系。結果表明,熱帶玉米染色體的SNP數目顯著的高于溫帶群體。熱帶玉米群體一共鑒定到14 123 408個SNP,染色體上平均每145個堿基存在一個變異位點;在溫帶玉米群體中共鑒定到個8 791 673個SNP,平均每234個堿基存在一個變異位點。溫帶和熱帶玉米群體一起進行SNP篩選,最終得到了204 752個符合過濾標準的SNP位點。此外,還分析了溫帶玉米和熱帶玉米群體中SNP的分布區域(表1),結果表明,溫帶和熱帶玉米群體中的SNP變異主要都發生在基因區間(Intergentic),此外依次是下游區間(Downstream)、上游區間(Upstream)、內含子區間(Intron)、外顯子區間(Exon)等。
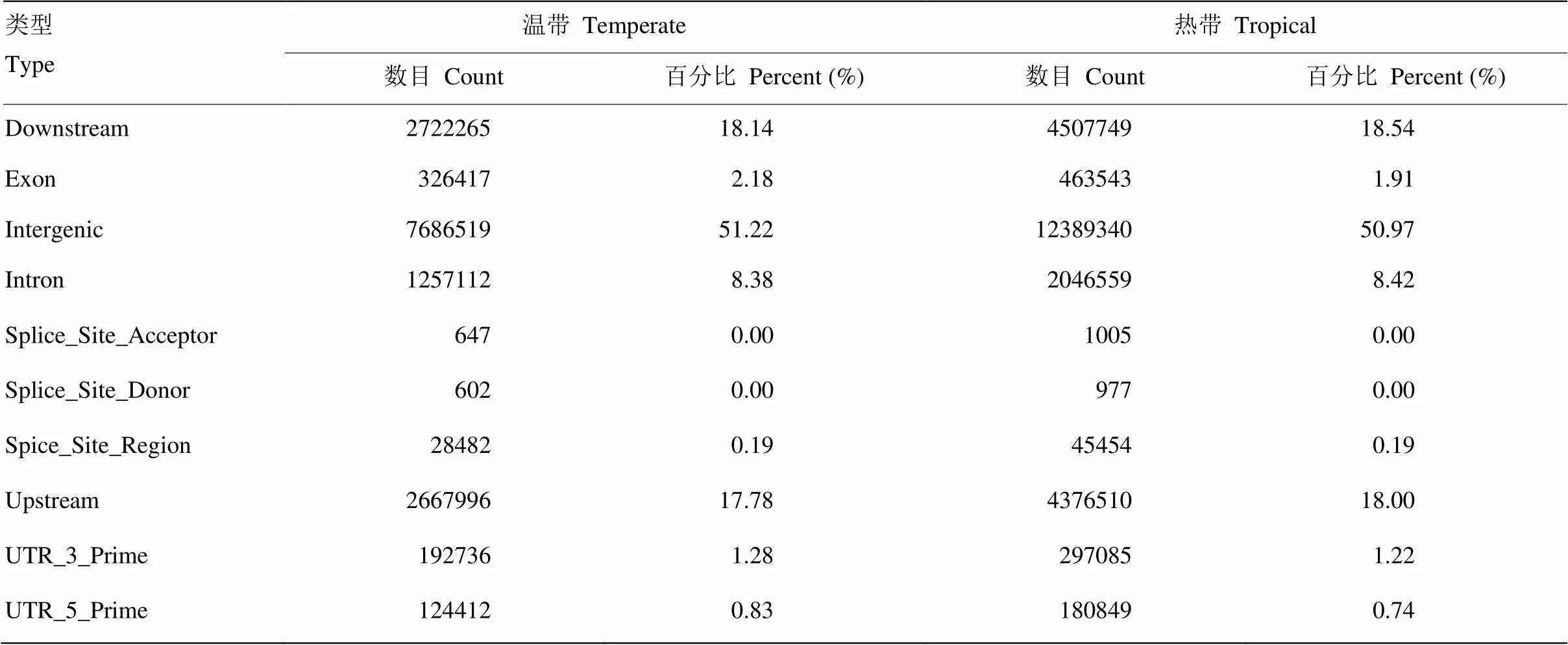
表1 溫帶和熱帶玉米群體的SNP在基因組區間的分布比例
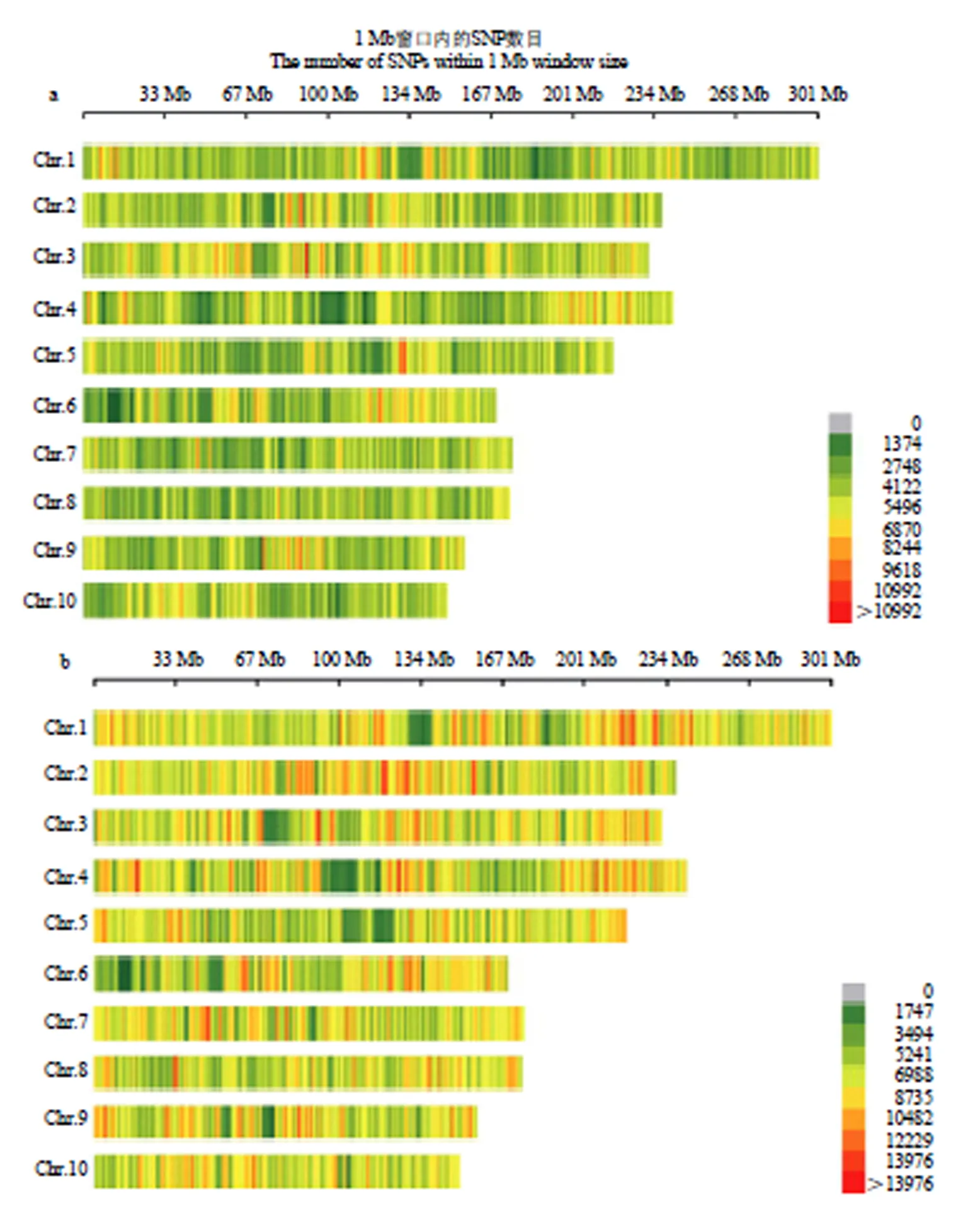
a:溫帶玉米群體SNP分布;b:熱帶玉米群體SNP分布。橫軸代表染色體的物理位置,窗口大小為1 Mb區間。深綠色代表SNP密度小的區域,紅色代表SNP密度高的區域
2.2 溫、熱帶玉米群體的主成分分析
溫帶玉米和熱帶玉米由于對光周期敏感性的不同,其表型存在較大差異(圖2),例如株高、葉片數、開花期等。溫帶材料可以在溫帶環境正常散粉開花,熱帶材料生殖生長期較長,未能在溫帶環境散粉開花。此外,熱帶材料的株高也顯著的高于溫帶材料。利用溫帶玉米和熱帶玉米基因組信息,借助主成分分析策略來揭示溫帶和熱帶玉米群體之間的遺傳關系(圖3),按照PC1和PC2 2個維度可以將使用的玉米自交系分為溫帶和熱帶2個類群。
2.3 FST值分析結果
溫帶玉米光周期敏感性低,可以在長日照的溫帶環境下正常生長結實;而熱帶玉米則具有較強的光周期敏感性,主要栽培于熱帶環境。利用2個群體共有的204 752個SNP標記計算得到每個滑動窗口內的F值,全基因組水平上F的top 1%是0.3593(圖4),高于閾值線的受選擇位點一共是1 908個,占總變異位點數的0.9%。其中第4染色體受到選擇的顯著性位點最多,是414個;第7染色體最少,是61個。第1染色體66 460 001—66 510 000區間內含有最高的F值,其值為0.77。在第1、2、3、4、5、6、8、9、10染色體上都有較強的選擇信號。

圖2 溫帶(a)和熱帶(b)玉米自交系表型
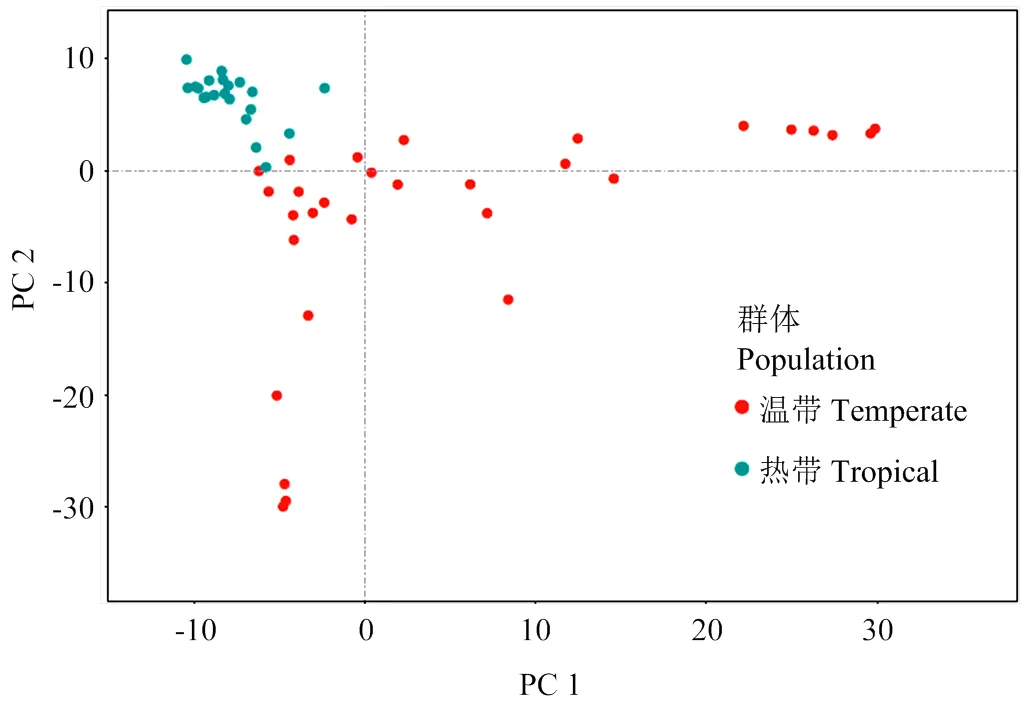
圖3 溫、熱帶群體的主成分分析
2.4 XP-EHH的分析
XP-EHH選擇信號檢測的原理是根據原有的突變區段由于連鎖效應較難被打破,而產生新的突變需要較長的選擇周期才能達到很高的基因頻率。因此,當某個單倍型區段在群體中出現頻率較高,代表該基因區段經歷了選擇,可以利用XP-EHH選擇信號的方法鑒定出來。利用selscan軟件計算全基因組的上每個SNP位點的XP-EHH值,使用溫帶群體作為試驗群體,熱帶群體作為參考群體,當XP-EHH值為正值時,代表在溫帶群體發生了選擇,其值為負值時,表明馴化選擇發生在熱帶群體。由于主要關注熱帶玉米向溫帶玉米的馴化,因此,只分析了XP-EHH值大于0的情況,即溫帶玉米群體中發生選擇的染色體區段(圖5),結果表明,在第2、3、7、8、10染色體存在較多的選擇區段,其top 1%為3.32,高于閾值線的SNP位點數目是39 664個,XP-EHH值最高的位點是位于Chr.2:69.324 Mb附近,其值為7.3298。以受選擇SNP位點為核心向上下游各擴增50 kb得到100 kb的候選基因區段,利用Bedtools軟件將其與玉米B73參考基因組進行比對,共得到1 913個候選基因,其中包含2個與玉米適應性改良關系密切相關的基因,分別是[33]和[34]。(Chr.9:115 786 897—115 789 787),其F值為3.58,最新的一項研究表明是玉米開花控通路中的一個重要調節因子[33],在長日照環境下,抑制開花促進因子[35-36]的表達,導致玉米晚花,通過CRISPR/Cas9技術介導的敲除,發現可以促進玉米提早開花。鑒定到在溫帶群體受到選擇,因而加速熱帶玉米群體對溫帶環境的適應,這與前人報道一致[33]。(Chr.9:108 447 974—108 449 794)是玉米光周期調控通路中的重要基因,該基因編碼蛋白可以和開花促進因子FT蛋白互作,加速玉米開花以適應長日照環境,是一個重要的開花調控基因。
2.5 候選基因的富集分析和功能注釋
利用F鑒定得到557個候選基因和XP-EHH得到的1 913個候選基因。基因富集分析表明F法得到的候選基因富集在生物學過程(biological process,P),共19個顯著的GO條目(表2)。這些GO條目主要涉及到肌動蛋白發育調控、微絲的發育調控和細胞骨架構成等。利用XP-EHH得到的1 913個基因沒有富集得到顯著的GO條目,推測是因為熱帶玉米馴化溫帶玉米涉及到眾多農藝性狀的改變,并且許多農藝性狀是由一系列微效數量位點控制,因此未能得到顯著的GO條目。盡管如此,XP-EHH的方法仍然可以得到較多的選擇基因,并且這些基因中鑒定得到了與玉米花期調控相關的基因,例如和。因此,通過選擇信號的方法可以為解析玉米馴化的遺傳機理、鑒定候選馴化基因提供實驗依據。除了這些已經報道過的調控基因,還統計了選擇信號值高于top 1%和top 0.1%的候選基因(表3)。另外,通過maizeGDB網站進行基因功能注釋,發現這些基因均是玉米生長發育過程中的重要調控因子,例如是一個光敏色素調節因子,參與玉米開花通路調控;參與下胚軸的發育調控。
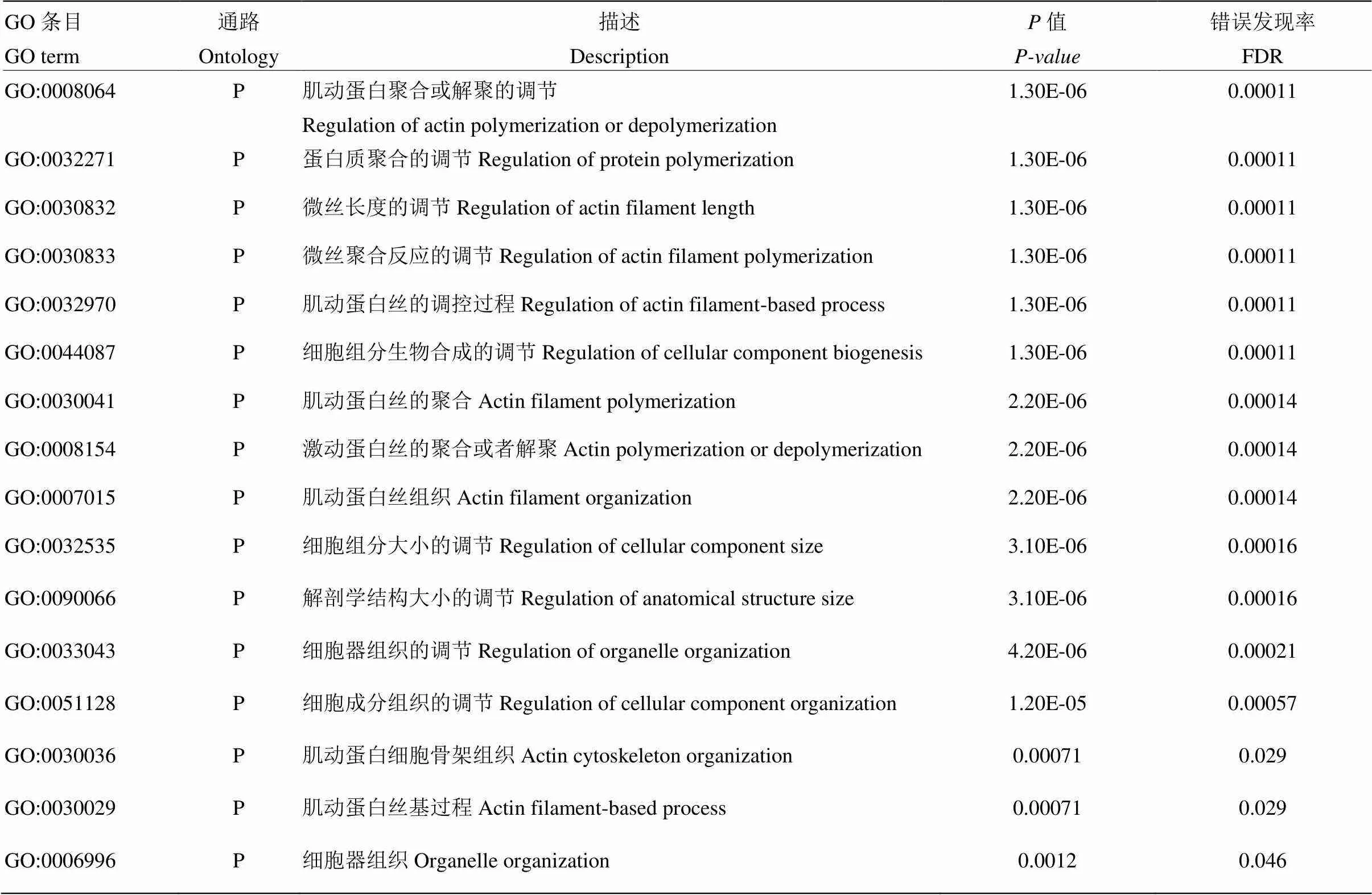
表2 FST選擇信號得到的基因進行GO富集分析的結果
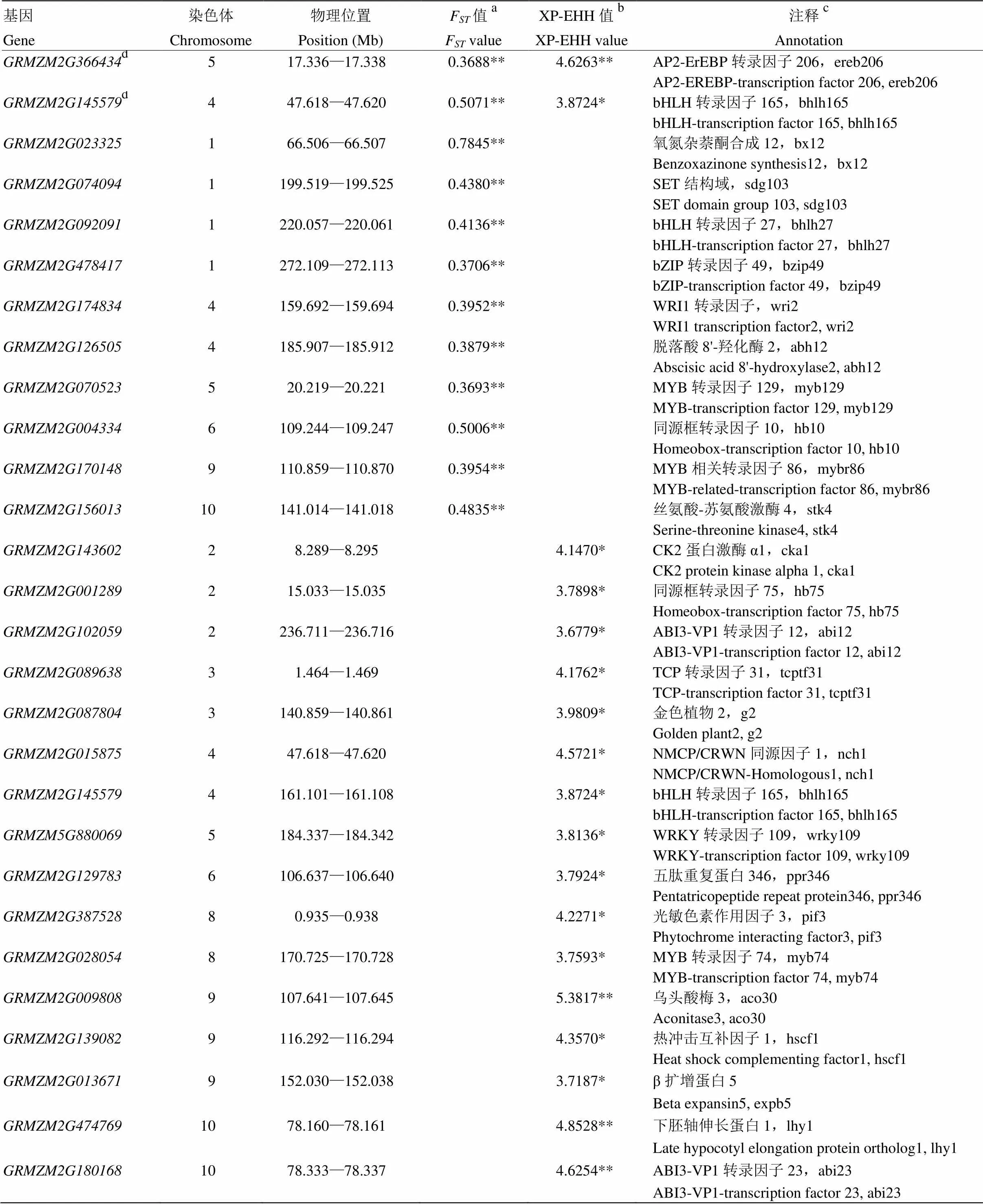
表3 選擇信號鑒定得到的候選基因功能注釋
a受選擇區域內較高F值的基因;b受選擇區域內較高XP-EHH值的基因;c基于maizeGDB的基因功能注釋;d同時受到F和XP-EHH選擇的基因;*代表受到選擇的關鍵基因(選擇信號值高于top 1%);**代表受到強烈選擇的基因(選擇信號值高于 top 0.1%)
aGenes with higherFvalues in selected regions;bGenes with higher XP-EHH values in selected regions;cGene function annotation based on maizeGDB;dCandidate genes identified by bothFand XP-EHH methods; *Represents the selected key genes (the selection signal over top 1%); **Represents the strongly selected genes (the selection signal over top 0.1%)
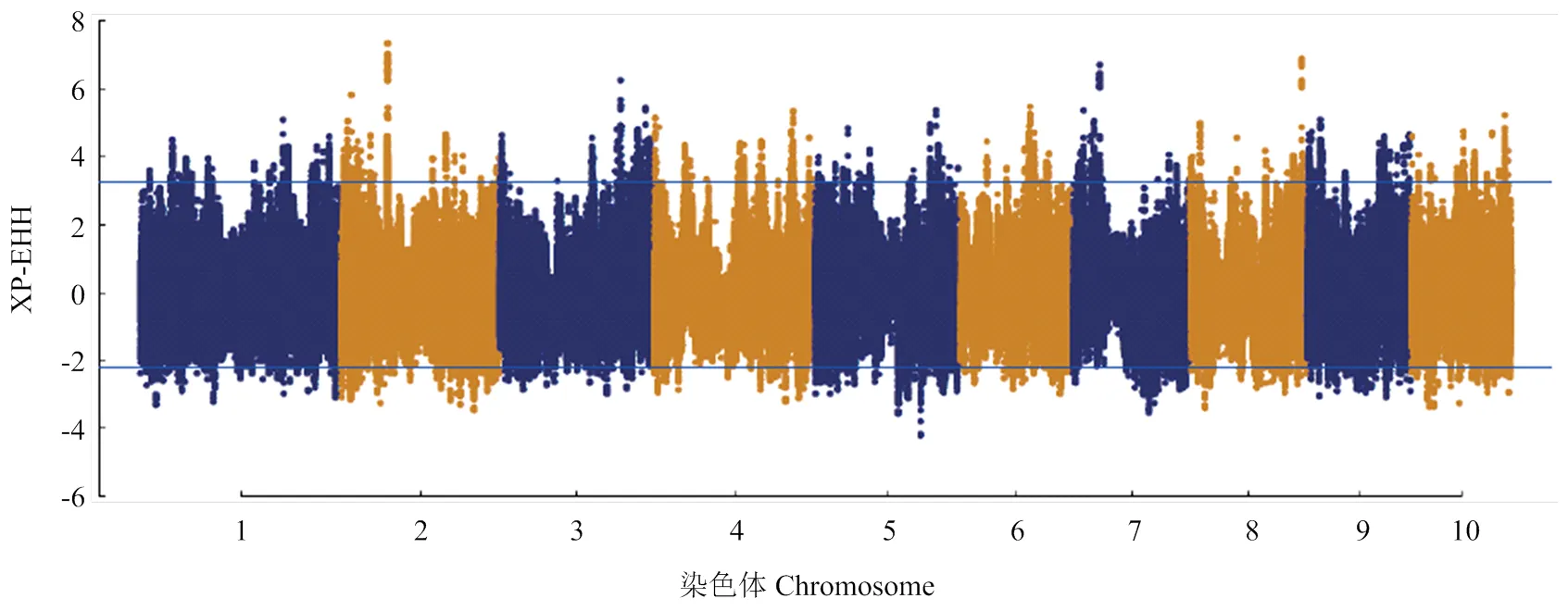
閾值線代表選擇信號值的top 1% the cutoff lines represent the top 1% of the XP-EHH selection signal
3 討論
3.1 溫熱帶玉米基因組變異分析
玉米起源于熱帶環境的墨西哥西南部,經過對其進行適應性改良,玉米逐漸的擴散到世界各地。根據光周期敏感性的不同,玉米可以被劃分為溫帶玉米和熱帶玉米[6]。熱帶玉米靠近玉米起源中心,很少經歷人工選擇和改良,保留著大量的有利變異,是進行群體遺傳改良的重要材料[8]。本研究選擇30份溫帶玉米自交系和21份熱帶玉米作為研究材料進行重測序。通過SnpEff軟件對變異文件進行基因組的功能的注釋和預測,發現熱帶玉米基因組具有更多的SNP(圖1),無論是SNP的總數還是平均分布密度均顯著高于溫帶玉米。該結果證明了熱帶玉米具有更高的遺傳多樣性,這與前人研究即熱帶玉米具有更高的遺傳多樣性一致[8]。主成分分析發現溫帶玉米群存在更大的變異幅度(圖2),推測是因為溫帶玉米群體包含較多的亞群,因此其變異幅度更廣,該結果和前人的主成分分析一致[6]。這也表明本研究所使用的基因型數據以及群體劃分的準確性,為后續的選擇信號分析奠定了數據基礎。玉米作為一個廣適性的作物,其在基因組水平存在多態性變化,進而導致相關表型變化,是使其成為有重要影響力作物的一個重要條件。研究揭示了溫熱帶玉米基因組水平SNP變異情況,可為后續的玉米功能基因組學研究提供理論基礎。
3.2 FST和XP-EHH的計算原理
隨著海量生物學數據的產出,使用高密度的SNP標記進行群體間選擇信號的分析在很多物種中都已成功進行,包括人類[37]、綿羊[38]、水稻[39]等。選擇信號是一種重要的群體遺傳學研究手段,利用合適的選擇信號可以顯著的揭示玉米適應性進化的遺傳機理,并且可以深入挖掘與農藝性狀相關的候選基因[21]。本研究利用F和XP-EHH的方法進行選擇信號的鑒定。F可以利用等位基因的頻率變化來篩選基因組受到馴化選擇的基因區段[16],這是因為同一個物種由于馴化的目的、種植環境、改良時間不同將會導致基因組上的等位基因頻率發生變化,雖然它可以鑒定出受到選擇的基因,但是不能確定選擇的方向[40],因此,本研究進一步引入XP-EHH選擇信號方法。XP-EHH是一種基于基因組上單倍型純合度的方法,鑒定不同群體間發生選擇作用的區段,XP-EHH統計分析方法引入了參考群體和試驗群體的概念,因此可以根據統計值判斷基因組發生選擇的方向,這為深入篩選候選馴化基因提供了更有力的數據支撐[41]。
3.3 選擇信號檢測結果的評價
傳統的選擇信號檢測方法主要利用RFLP、AFLP和SSR等分子標記。有研究利用玉米和大芻草的99個SSR標記作為選擇信號進行分析[2],揭示了現代玉米起源于約9 000年前的墨西哥西南部。然而RFLP、AFLP和SSR等分子標記開發程序繁瑣,并且定位精度不高,如今已經很少應用于選擇信號的研究之中。隨著新一代測序技術的發展,基于高通量測序技術得到的SNP標記由于其精度高、密度大以及定位更加準確等優點,逐漸取代了傳統的分子標記,成為進行選擇信號檢測的主要方法[19-21]。本研究選擇具有代表性的溫帶和熱帶玉米群體的重測序數據,進行全基因組水平的F和XP-EHH的2種選擇信號的鑒定,分別篩選到了557和1 913個候選基因。利用F鑒定得到的基因進行富集分析,結果表明這些基因富集在一些重要的生物學通路上,主要參與肌動蛋白的發育調節(GO:0008064,GO:0032956)、微絲的發育調節(GO:0030832)、細胞骨架發育調控(GO:0030036)等。本研究利用F得到基因進行富集分析,得到的顯著GO條目較少,此外XP-EHH選擇信號法鑒定到的候選基因未能得到顯著的GO條目,不能準確地反映群體分化的結果。推測是目前所用的參考數據庫信息不夠全面,因此未能特異的鑒定出富有生物學意義的GO條目。本研究通過對具有較高選擇信號的基因進行功能注釋,發現這些基因是在熱帶玉米改良為溫帶玉米過程中受到了極大的選擇,同時也是玉米生長發育過程中重要的調控因子(表3)。因此,結合相關的表型性狀和選擇信號法可以鑒定玉米適應改良過程中發生變化的基因。其中通過XP-EHH選擇信號法,鑒定到溫熱帶玉米適應性改良相關的調控基因[33]和玉米開花調控相關的基因[34]。盡管前人曾經利用過相關的選擇信號鑒定了玉米在馴化過程中的選擇基因[21],然而本研究著重挖掘了與開花相關的調控基因,開花是玉米生育期的一個重要農藝性狀,關系到植株從營養生長到生殖生長的轉變。其中包括已經報道過的和。此外在候選基因集中鑒定到了一些開花調控候選基因,例如,基因功能注釋表明該基因是一個光敏色素互作因子(表3),與光周期基因互作[42]。因此,本研究鑒定得到的候選基因可以為后續的開花遺傳機理解析提供理論基礎。這些研究表明結合F和XP-EHH選擇信號法是進行群體遺傳分化、功能富集分析和馴化基因挖掘的一個重要手段。
4 結論
基于對溫帶和熱帶玉米的基因型數據分析,發現熱帶玉米具有更高的遺傳多樣性,表明其是玉米分子育種的重要資源。鑒定到玉米馴化改良過程中受到選擇的基因,且部分受選擇的基因參與玉米開花發育調控途徑。
[1] PIPERNO D R, RANERE A J, HOLST I, IRIARTE J, DICKAU R. Starch grain and phytolith evidence for early ninth millennium BP maize from the Central Balsas River Valley, Mexico., 2009, 106(13): 5019-5024.
[2] MATSUOKA Y, VIGOUROUX Y, GOODMAN M M, SANCHEZ J, BUCKLER E, DOEBLEY J. A single domestication for maize shown by multilocus microsatellite genotyping., 2002, 99(9): 6080-6084.
[3] VAN HEERWAARDEN J, DOEBLEY J, BRIGGS W H, GLAUBITZ J C, GOODMAN M M, Gonzalez J d J S, ROSS-IBARRA J. Genetic signals of origin, spread, and introgression in a large sample of maize landraces., 2011, 108(3): 1088-1092.
[4] BUCKLER E S, HOLLAND J B, BRADBURY P J, ACHARYA C B, BROWN P J, BROWNE C, ERSOZ E, FLINT-GARCIA S, GARCIA A, GLAUBITZ J C, et at.. The genetic architecture of maize flowering time., 2009, 325(5941): 714-718.
[5] SWARTS K, GUTAKER R M, BENZ B, BLAKE M, BUKOWSKI R, HOLLAND J, KRUSE-PEEPLES M, LEPAK N, PRIM L, ROMAY M C, et at. Genomic estimation of complex traits reveals ancient maize adaptation to temperate North America., 2017, 357(6350): 512-515.
[6] LU Y L, YAN J B, GUIMARAES C T, TABA S, HAO Z F, GAO S B, CHEN S J, LI J S, ZHANG S H, VIVEK B S, et at.. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms., 2009, 120(1): 93-115.
[7] TALLURY S, GOODMAN M. Experimental evaluation of the potential of tropical germplasm for temperate maize improvement., 1999, 98(1): 54-61.
[8] HALLAUER A R, CARENA M J. Adaptation of tropical maize germplasm to temperate environments., 2013, 196(1): 1-11.
[9] KIMURA M. Evolutionary rate at the molecular level., 1968, 217(5129): 624-626.
[10] SMITH J M, HAIGH J. The hitch-hiking effect of a favourable gene., 1974, 23(1): 23-35.
[11] TAJIMA F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism., 1989, 123(3): 585-595.
[12] NIELSEN R, WILLIAMSON S, KIM Y, HUBISZ M J, CLARK A G, BUSTAMANTE C. Genomic scans for selective sweeps using SNP data., 2005, 15(11): 1566-1575.
[13] SABETI P C, REICH D E, HIGGINS J M, LEVINE H Z, RICHTER D J, SCHAFFNER S F, GABRIEL S B, PLATKO J V, PATTERSON N J, MCDONALD G J, et at.. Detecting recent positive selection in the human genome from haplotype structure., 2002, 419(6909): 832.
[14] VOIGHT B F, KUDARAVALLI S, WEN X Q, PRITCHARD J K. A map of recent positive selection in the human genome., 2006, 4(3): e72.
[15] SABETI P C, VARILLY P, FRY B, LOHMUELLER J, HOSTETTER E, COTSAPAS C, XIE X, BYRNE E H, MCCARROLL S A, et at.. Genome-wide detection and characterization of positive selection in human populations., 2007, 449(7164): 913.
[16] WRIGHT S. The genetical structure of populations., 1949, 15(1): 323-354.
[17] WEIR B S, COCKERHAM C C. Estimating F-statistics for the analysis of population structure., 1984, 38(6): 1358-1370.
[18] GIANOLA D, SIMIANER H, QANBARI S. A two-step method for detecting selection signatures using genetic markers., 2010, 92(2): 141-155.
[19] AXELSSON E, RATNAKUMAR A, ARENDT M L, MAQBOOL K, WEBSTER M T, Perloski M, Liberg O, ARNEMO J M, HEDHAMMAR A, LINDBLAD-TOH K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet., 2013, 495(7441): 360.
[20] LIU K J, GOODMAN M, MUSE S, SMITH J S, BUCKLER E, DOEBLEY J. Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites., 2003, 165(4): 2117-2128.
[21] HE C, FU J J, ZHANG J, Li Y X, ZHENG J, ZHANG H W, YANG X H, WANG J H, WANG G Y. A gene-oriented haplotype comparison reveals recently selected genomic regions in temperate and tropical maize germplasm., 2017, 12(1): e0169806.
[22] SCHNABLE P S, WARE D, FULTON R S, STEIN J C, WEI F S, PASTERNAK S, LIANG C Z, ZHANG J W, FULTON L, GRAVES T A, et at.. The B73 maize genome: complexity, diversity, and dynamics., 2009, 326(5956): 1112-1115.
[23] JIAO Y P, PELUSO P, SHI J H, LIANG T, STITZER M C, WANG B, CAMPBELL M S, STEIN J C, WEI X H, CHIN C S, et at.. Improved maize reference genome with single-molecule technologies., 2017, 546(7659): 524.
[24] CHIA J M, SONG C, BRADBURY P J, COSTICH D, DE LEON N, DOEBLEY J, ELSHIRE R J, GAUT B, GELLER L, GLAUBITZ J C, et at.. Maize HapMap2 identifies extant variation from a genome in flux., 2012, 44(7): 803.
[25] BUKOWSKI R, GUO X S, LU Y L, ZOU C, HE B, RONG Z Q, WANG B, XU D W, YANG B C, XIE C X, et at.. Construction of the third-generation Zea mays haplotype map., 2017, 7(4): gix134.
[26] DANECEK P, AUTON A, ABECASIS G, ALBERS C A, BANKS E, DEPRISTO M A, HANDSAKER R E, LUNTER G, MARTH G T, SHERRY S T, et at.. 1000 Genomes Project Analysis Group. The variant call format and VCFtools., 2011, 27(15): 2156-2158.
[27] CINGOLANI P, PLATTS A, WANG L L, COON M, NGUYEN T, WANG L A, LAND S J, LU X Y, RUDEN D M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of drosophila melanogaster strain w1118; iso-2; iso-3., 2012, 6(2): 80-92.
[28] MA Y, DING X, QANBARI S, WEIGEND S, ZHANG Q, SIMIANER H. Properties of different selection signature statistics and a new strategy for combining them., 2015, 115(5): 426.
[29] HOAGLIN D C, MOSTELLER F, TUKEY J W.. New York: John Wiley & Sons, 1983.
[30] SZPIECH Z A, HERNANDEZ R D. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection., 2014, 31(10): 2824-2827.
[31] QUINLAN A R, HALL I M. BEDTools: a flexible suite of utilities for comparing genomic features., 2010, 26(6): 841-842.
[32] TIAN T, LIU Y, YAN H Y, You Q, Yi X, Du Z, XU W Y, SU Z. agriGO v2. 0: a GO analysis toolkit for the agricultural community, 2017 update., 2017, 45(W1): W122-W129.
[33] HUANG C, SUN H Y, XU D Y, LIANG Y M, WANG X F, XU G H, TIAN J G, WANG C L, LI D, WU L H, et at..enhances maize adaptation to higher latitudes., 2018, 115(2): E334-E341.
[34] KHAN S, ROWE S C, HARMON F G. Coordination of the maize transcriptome by a conserved circadian clock., 2010, 10(1): 126.
[35] GUO L, WANG X, ZHAO M, HUANG C, LI C, LI D, YANG C , YORK A M, XUE W, XU G, LIANG Y, CHEN Q, DOEBLEY J F, TIAN F. Stepwise cis-regulatory changes incontribute to maize flowering-time adaptation., 2018, 28(18): 3005-3015.
[36] MENG X, MUSZYNSKI M G, DANILEVSKAYA O N. The-likegene functions as a floral activator and is involved in photoperiod sensitivity in maize., 2011, 23(3): 942-960.
[37] SHEEHAN M J, NACHMAN M W. Morphological and population genomic evidence that human faces have evolved to signal individual identity., 2014, 5: 4800.
[38] 曾滔, 趙福平, 王光凱, 吳明明, 魏彩虹, 張莉, 李利, 張紅平, 杜立新. 基于群體分化指數FST的綿羊全基因組選擇信號檢測. 畜牧獸醫學報, 2013, 44(12): 1891-1899.
ZENG T, ZHAO F P, WANG G K, WU M M, WEI C H, ZHANG L, LI L, ZHANG H P, DU L X. Genome-wide detection of selection signatures in sheep populations with use of population differentiation index FST., 2013, 44(12): 1891-1899. (in Chinese)
[39] HUANG X, SANG T, ZHAO Q, QI F, ZHAO Y, LI C Y ZHU C R, LU T T, ZHANG Z W, LI M, et at.. Genome-wide association studies of 14 agronomic traits in rice landraces., 2010, 42(11): 961.
[40] MCVICKER G, GORDON D, DAVIS C, GREEN P. Widespread genomic signatures of natural selection in hominid evolution., 2009, 5(5): e1000471.
[41] 薛周艤源, 宋顯威, 吳林慧, 王露珍, 崔家安, 孫章健, 張政, 馬云龍. 畜禽選擇信號檢測方法及其統計學問題. 畜牧獸醫學報, 2018, 49(6): 1099-1107.
XUE Z Y Y, SONG X W, WU L H, WANG L Z, CUI J A, SUN Z J, ZHANG Z, MA Y L. The identification methods of selection signatures in livestock and its statistical problems., 2018, 49(6): 1099-1107. (in Chinese)
[42] KUMAR I, SWAMINATHAN K, HUDSON K, HUDSON M E. Evolutionary divergence of phytochrome protein function inPIF3 signaling., 2016, 67(14): 4231-4240.
(責任編輯 李莉)
Genome-Wide Detection of Selection Signal in Temperate and Tropical Maize Populations with Use ofFand XP-EHH
YANG YuXin, ZOU Cheng
(Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing 100081)
【Objective】Maize was first domesticated in tropical areas, but it has been cultivated widely in the temperate regions after natural and artificial selection. Flowering time is not only the key component of the entire growth period, but also a major adaptive trait during the dispersal process from tropical to temperate conditions. Thus, identifying the selected gene regions responsible for the adaptation to temperate zones, and discovering the genes that are involved in flowering time could provide a molecular basis for improving maize and for dissecting its flowering mechanism. 【Method】We analyzed the haplotype data of 30 temperate and 21 tropical maize inbred lines. High quality SNP (single nucleotide polymorphism) markers were obtained after filtering out SNPs with high missing rates and low allele frequencies. These high quality SNPs were annotated by SNPeff. Principle component analysis (PCA) of the genotypic data of temperate and tropical maize was performed to further validate the population structure of these samples. Using high quality SNP markers that were present in tropical and temperate populations, we calculated the selection signal using the fixation index (F) and cross population extended haplotype homozygosity (XP-EHH) methods. The top 1% of values was used as a significant threshold to identify the candidate selected signals. The candidate selected genes that we selected from temperate and tropical maize were identified based on their SNP annotation. The function of these selected genes was characterized furtherly by the GO enrichment analysis using agriGO. To identify the genes for flowering time that were under selection, bioinformatics databases were examined that contained relevant data on maize. 【Result】By analyzing the high depth resequencing data, we found 14123408 and 8791673 SNPs in tropical and temperate populations, respectively. The identified SNPs were mainly distributed in the intergenic regions. There were 204752 high quality SNPs that coexisted in temperate and tropical populations. PCA indicated that temperate and tropical maize can be divided into two groups. The top 1% ofFvalue and XP-EHH were 0.3059, 3.2681, and a total of 557 and 1 913 candidate genes were identified byFand XP-EHH methods, respectively. Many candidate genes were highly related to regulation of flowering time, which included,and.is a vital gene for regulating flowering time, and it negatively regulated the floral activator gene, which cause the late flowering time phenotype under long-day conditions. COL1 positively interacts with the FT protein to promote the transition of flowering time to adapt to the long-day environment. Functional annotations ofrevealed that it was a phytochrome interacting factor, and interacts with photoperiod gene. 【Conclusion】Our study revealed that tropical maize had higher genetic diversity than temperate maize. A series of genes that were under selection during the adaptation to tropical to temperate conditions were predicted, and we further explored the genes that were involved in flowering during this process.
maize; selection signal; fixation index; cross population extended haplotype homozygosity; flowering time genes
2018-10-30;
2018-12-09
國家重點研發計劃(2016YFD0100303)、國家自然科學基金面上項目(31371638)
楊宇昕,E-mail:yyx0719@126.com。通信作者鄒棖,E-mail:zoucheng@caas.cn
10.3864/j.issn.0578-1752.2019.04.001
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
