Ce和O空位共摻雜TiO2的電子結構與光學性質*
2019-03-13 03:03:34周詩文彭平陳文欽庾名槐郭惠袁珍
物理學報 2019年3期
關鍵詞:實驗
周詩文 彭平 陳文欽 庾名槐 郭惠 袁珍
1) (海南大學材料與化工學院, 海口 570228)
2) (湖南大學材料科學與工程學院, 長沙 410082)
(2018 年 11 月 1 日收到; 2018 年 12 月 11 日收到修改稿)
采用基于密度泛函理論加U的計算方法, 研究了Ce和O空位單(共)摻雜銳鈦礦相TiO2的電子結構和光吸收性質. 計算結果表明, Ce和O空位共摻雜TiO2的帶隙中出現了雜質能級, 且帶隙窄化為2.67 eV, 明顯比純TiO2和Ce, O空位單摻雜TiO2的要小, 因而可提高TiO2對可見光的響應能力, 使TiO2的光吸收范圍增加. 光吸收譜顯示, 摻雜后TiO2的光吸收邊發生了顯著紅移; 在400.0—677.1 nm的可見光區, 共摻雜體系的光吸收強度顯著高于純TiO2和Ce單摻雜TiO2, 而略低于O空位單摻雜TiO2. 此外, Ce摻雜TiO2中引入O空位后, TiO2的導帶邊從?0.27 eV變化為?0.32 eV, 這表明TiO2的導帶邊的還原能力得到了加強.計算結果為Ce和O空位共摻雜TiO2在可見光光解水方面的進一步研究提供了有力的理論依據.
1 引 言
1972 年, Fujishima 和 Honda[1]首次報道了在紫外光的照射下TiO2可以使水發生水解, 從而掀起了對TiO2光催化性能的研究熱潮. 最近幾十年來, 人們從光解水[1,2]、太陽能電池[3]、自清潔[4]、污染物降解[5]等方面對TiO2的應用進行了廣泛而深入的研究. 這些研究表明, TiO2具有高氧化能力、良好的化學穩定性及無毒、廉價等優異性能[6,7], 是一種較理想的光催化半導體, 在環保和能源等領域極具應用潛力. 然而, TiO2在光催化和能量轉換應用過程中仍存在兩大亟待解決的瓶頸[8]: 一是, 由于 TiO2的本征帶隙較寬 (約 3.0 eV), 純 TiO2只能吸收占太陽光譜約5%、波長小于387.5 nm的紫外光, 卻不能響應占太陽光約43%的可見光; 二是, TiO2材料中光生載流子的復合率較高, 光生電子-空穴對難以有效分離, 致使TiO2的光量子產率偏低. 為此, 人們利用離子摻雜、貴金屬沉積、半導體復合、染料敏化, 以及基于能帶工程設計的異質結等方法和技術[9,10]來提高TiO2材料對太陽光能的利用率與光催化活性.
最近, 許多實驗和理論研究均發現鑭系離子在裁剪TiO2的能帶結構和增強TiO2的光催化活性方面具有特殊的作用. 如, Mazierski等[11]利用電化學方法制備了一系列的 Er-, Yb-, Ho-, Gd-, Pr-摻雜的TiO2納米管, 發現這些鑭系離子摻雜均能增強TiO2的光催化活性. 在可見光解降甲苯的過程中, Ho摻雜TiO2的效率尤為顯著; 進一步的理論計算表明, 能帶中新出現的Ho-f能級和表面缺陷態可能是TiO2可見光催化能力增強的主要原因.Du等[12]報道Ce摻雜TiO2體系不但展現出優異的可見光吸收活性, 而且顯示出良好的熱穩定性.Nasir等[13]認為這種光催化活性增強的原因之一在于TiO2中形成了氧化還原電荷對(即Ce3+/Ce4+,其中Ce3+具有4f15d0電子構型, 而Ce4+具有4f05d0電子構型), 因而在光生載流子的過程中Ce3+/Ce4+充當了“清道夫”的作用, 有效促進了電子-空穴對的分離, 提高了 TiO2的光催化活性. Li等[14]發現,當TiO2表面的Ce/Ti原子的比率為2.0%時, 在可見光照射下Ce-TiO2顯示出優良的光催化活性.Xiao等[15]指出隨著Ce摻雜濃度的提高, TiO2納米纖維的光吸收邊從450.0 nm大幅紅移至739.0 nm,當Ce/Ti的摩爾比增加到0.03后, 其可見光的吸收能力又逐漸變小. 而Fu等[16]通過第一性原理計算則發現, 隨著 Ce 摻雜濃度的增加, Ti 3d 軌道電子在導帶底的態密度不斷減弱, 從而使TiO2在180.0—600.0 nm之間的光吸收強度逐漸降低. 可見, 有關Ce摻雜TiO2的光吸收性能的理論研究與實驗研究之間仍存在一定的差距. 這些差距的形成與多方面原因密切相關. 例如, 實驗制備的摻雜體系, 不可避免地會產生一些缺陷(如氧空位(OV)、間隙摻雜、異種原子置換摻雜等缺陷), 而理論計算中常常卻忽略了某些缺陷, 因而計算結果與實驗觀測之間有一定的差距. 對于金屬氧化物而言, OV是一種常見的缺陷, 對金屬氧化物的電子結構和物化性能可產生重要影響[17]. 最近, 占昌朝等[18]報道 La3+置換 TiO2中的 Ti4+后形成了 OV,從而引起刃型位錯, 他們認為這些位錯缺陷為光催化反應活性位點提供了可能, 十分有利于增強TiO2的光催化活性. 再如, 最近 Yang 等[19]研究發現: 當氫原子占據OV, 形成新型的OVH缺陷時,銳鈦礦相TiO2的能帶中出現了新的亞能級, 從而有效提高了TiO2的可見光吸收能力; 利用可見光輻射含OVH缺陷的TiO2光陽極, 可以把水完全裂解.
綜上所述, 離子摻雜和空位缺陷均對氧化物半導體的能帶結構、光吸收性能以及光催化活性具有重要的調控與影響作用. 鑒于Ce與OV共摻雜TiO2的理論和實驗研究鮮有報道, 本文對Ce/OV共摻雜銳鈦礦相TiO2的電子結構和光學性質進行理論計算和研究.
2 物理模型與計算方法
銳鈦礦型TiO2晶體結構屬四方晶系, 空間群為I41/amd, 每個晶胞內包含12個原子, 由 4個TiO2基本單元構成. 其中, Ti原子處于由鄰近6個O原子所圍成的八面體中心, Ti原子的配位數為6, O原子配位數為3, 而且O原子的位置只與一個內坐標u有關, Ti和O兩種原子的坐標分別為 Ti(0, 0, 0)和 O(0, 0,u). 已有諸多理論研究[8,16,20,21]表明采用 2 × 2 × 1 的銳鈦礦相 TiO2超胞結構進行周期性計算, 可獲得可靠的計算結果,此時相鄰超胞中摻雜原子間的相互作用可以忽略.另一方面, 采用 2 × 2 × 1 超胞結構, 摻雜濃度可調控到與最近許多實驗和理論研究中相近的摻雜濃度[12?14,20]. 考慮到以上兩個因素, 本研究各計算模型均采用 2 × 2 × 1 的超胞結構 (各超胞模型的原子和缺陷位置如圖1所示). 其中, 純銳鈦礦相TiO2, Ce摻雜 TiO2, 含 OV的 TiO2, 以及 Ce與OV共摻雜 TiO2的結構模型分別命名為: pure TiO2, Ce-TiO2, OV-TiO2, Ce/OV-TiO2.
本文采用Materials Studio軟件中基于密度泛函理論 (density functional theory, DFT) 的平面波超軟贗勢法的CASTEP程序包進行計算. 考慮到計算體系含有強關聯性的局域性Ti 3d和Ce 4f電子, 采用標準的DFT計算難以獲得滿意的電子結構和光學性質. 因此, 計算過程中交換關聯泛函

圖1 2 × 2 × 1 的超胞結構圖 (紅色球為 O, 灰色球為Ti, 藍色球表示Ce原子的摻雜位置, 紅色虛圓圈表示OV 的 位 置 ) (a) Pure TiO2; (b) Ce-TiO2; (c) OV-TiO2;(d) Ce/OV-TiO2Fig.1. The 2 × 2 × 1 supercell structure of various supercell models (the red, grey and blue balls represent O,Ti and Ce atoms, respectively; the imaginary red circle shows the position of oxygen vacancy): (a) Pure anatase TiO2; (b) Ce-TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2.
采用局域密度近似 (local density approximation,LDA), 并對具有強關聯作用的 Ti 3d 和 Ce 4f電子用U值修正.U值修正[22]是指對強定域性的電子 (如 d, f軌道電子), 引入一個在位 (on-site)的庫侖排斥項U, 從而修正交換關聯泛函對強關聯電子局域性的低估, 以達到合理的結果. 基于文獻[16, 21]提供的U值, 并通過對 TiO2和 CeO2的帶隙與U之間的關系測試發現: 當取UTi 3d =UCe 4f =8.0 eV 時, TiO2和 CeO2的帶隙值分別為 3.12 和2.78 eV. 此測試結果與采用相同U值的理論計算結果[16,23]吻合極好; 與實驗測量值[24,25]也非常接近;且測試過程中, TiO2和CeO2晶格參數相對于初始的參數變化均小于2.0%, 計算得到的電子能帶結構也可靠合理. 因此, 計算中 Ti 3d 和 Ce 4f軌道的U值均設為 8.0 eV.
計算中平面波截斷動能設置為620 eV, 采取6 × 6 × 5 的 Monkhorst-Pack 格點取樣. 自洽場運算中應用Pulay密度混合法, 能量收斂標準為1.0 × 10?5eV/atom (1 eV/atom = 9.6485 × 104J/mol), 最大公差為 0.0002 nm, 晶體內應力收斂標準為0.05 GPa, 每個原子間相互作用力小于0.2 eV/nm. 計算電子能帶結構和光學性質之前,計算所用的超胞結構先采用BFGS (Broyden-Flecher-Goldfarb-Shanno)方法進行幾何優化, 然后再對優化后的模型結構進行單點能計算. 參與計算的價電子的構型分別為 O: 2s22p4, Ti: 3s23p63d24s2,Ce: 4f15s25p65d16s2. 為保證計算結果的可比性, 各模型的計算均在倒易空間中進行, 其參數的設置均與上述設置相同.
為便于和實驗測量結果比較, 光吸收性質計算中運用了非極化多晶模型, 并利用剪刀算符修正帶隙值. 考慮到純 TiO2的實驗帶隙值為 3.23 eV, 而本計算的值為 3.12 eV, 兩者間相差 0.11 eV. 因此,剪刀算符設為0.11 eV. 在能帶帶邊的計算中亦使用0.11 eV對各體系的帶隙寬度進行修正.
3 結果與討論
3.1 結構優化結果
表1為采用廣義密度梯度近似(generalized gradient approximation, GGA), GGA +U, LDA及LDA +U方法, 對純TiO2進行結構優化及能帶結構計算得到的晶格參數和帶隙值. 其中, 采用LDA +U計算得到的純TiO2的晶格常數和內坐標參數 (a,b,c,u) (u=dap/c)的計算誤差均小于2.0%, 與實驗和理論計算值[16,20,21,24,26?30]均吻合良好. 表 2 為采用 LDA +U方法, 對 Ce/OV單 (共)摻雜TiO2的超晶胞結構幾何優化后, 折合成單晶胞的結構參數.
表2數據表明, 摻雜體系的晶格參數均發生了不同程度的畸變. 由晶體場理論可知[11]: 晶格畸變可導致TiO2中八面體單元的正負電荷中心分離,從而在晶體中形成局域內建電場. 由于產生了內建電場, TiO2中光生電子-空穴對的分離和遷移變得更加容易, 十分有利于增強TiO2的光催化活性.這種內建電場的強弱, 可以用八面體單元的電偶極矩來度量. 表3列出了摻雜體系的平均電偶極矩的計算值.
表3中平均偶極矩的計算結果表明: OV可顯著提高TiO2中八面體結構的平均偶極矩; Ce摻雜TiO2中引入OV后, 八面體結構單元的平均電偶極矩的大小明顯增加. 可見, 形成一定濃度的OV, 有利于增強TiO2的光催化活性. 后文中的電子能帶結構、態密度和光吸收譜的計算結果均進一步印證了這一結論.時,μO由 2μO=μTiO2-μTi決定. Ce 的化學勢由關系式μCe=μCeO2-μO2獲得. 各摻雜體系在富氧和富鈦條件下的形成能的計算結果列于表3. 結果表明, OV-TiO2在富氧和富鈦條件下的形成能的大小分別與Wu等[33]在富氧和富鈦條件下的計算值1.00和5.30 eV極其接近. 比較共摻雜與單摻雜體系的形成能大小可知: Ce/OV共摻雜TiO2的缺陷形成能顯著低于單摻雜TiO2的形成能, 負的形成能說明Ce/OV共摻雜體系具有良好的穩定性.
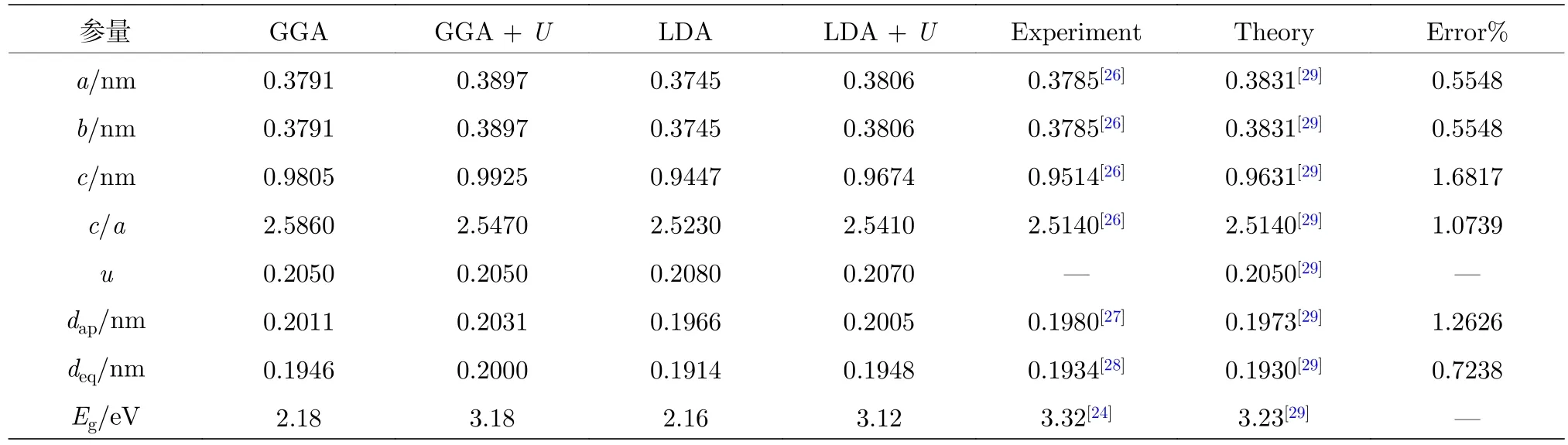
表1 采用 GGA, GGA + U, LDA 及 LDA + U 方法計算得到的純銳鈦礦相 TiO2 的晶格參數和帶隙寬度Table 1. Lattice parameters and band gap width of pure anatase TiO2 calculated at the GGA, GGA + U, LDA and LDA + U levels of theory.
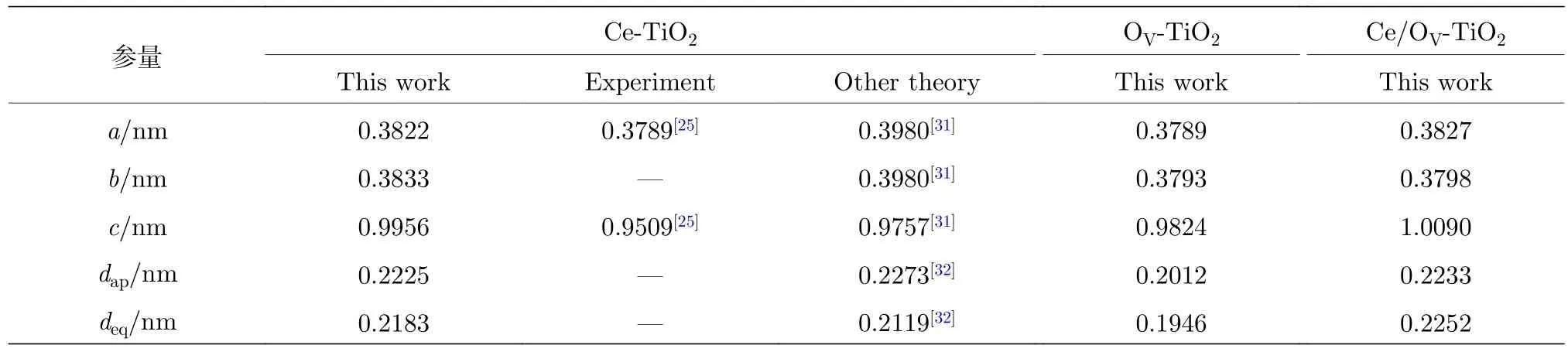
表2 Ce/OV 單 (共)摻雜銳鈦礦相 TiO2 弛豫后的晶胞參數Table 2. Lattice parameters of Ce/OV single and co-doped anatase TiO2 after the structure relaxation.
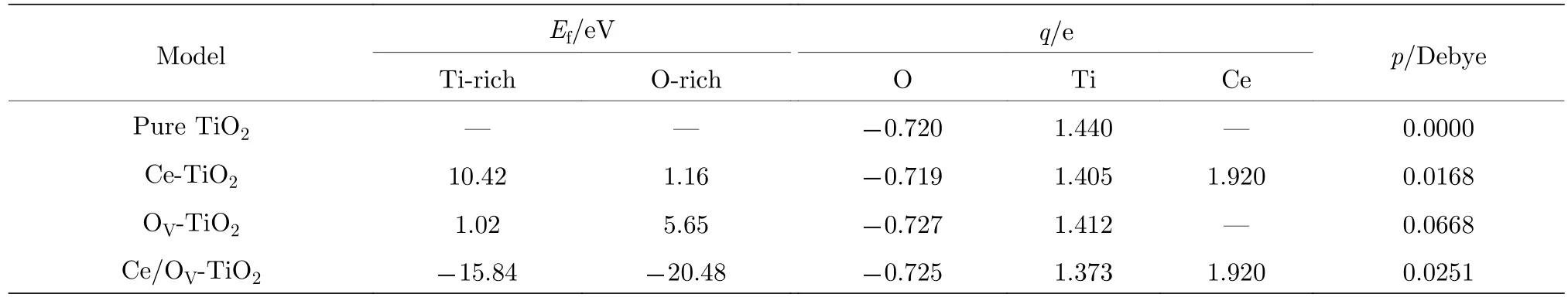
表3 Ce/OV摻雜銳鈦礦相TiO2的缺陷形成能(Ef)、平均凈電荷(q)和平均偶極矩(p)Table 3. Defect formation energy, average net charges and average dipole moment of Ce/OV doped TiO2 supercell after the structure relaxation.
3.2 缺陷形成能
為評估摻雜原子嵌入TiO2晶格與空位形成的相對難易程度, 以及確定摻雜體系的穩定性, 這里采用缺陷形成能來表征[20,29]. 缺陷形成能(Ef)的計算公式如下:
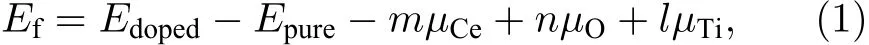
其中,Edoped與Epure分別代表摻雜(缺陷)模型與純 TiO2超胞模型的總能量;μCe,μO,μTi分別表示 Ce原子、O 原子、Ti原子的化學勢;m,n,l分別表示摻雜進入TiO2的Ce原子的個數, 以及從TiO2中移除的O原子和Ti原子的個數. 對于Ce-TiO2模型,m= 1,n= 0,l= 1; 類似地, OVTiO2模型,m= 0,n= 1,l= 0; Ce/OV-TiO2模型,m= 1,n= 1,l= 1. 眾所周知, 材料的缺陷形成能與其生長制備條件密切相關. 因此, 本文只計算富氧(O-rich)和富鈦(Ti-rich)條件下的形成能.
在富氧條件下, 顯然,μO=μO2/2 (μO2為氧氣分子的總能); 此時,μTi的大小則由關系式μTiO2=μTi+2μO得出. 在富鈦條件下,μTi的大小取自密排六方相(hcp)α-Ti金屬中單個原子的能量; 此
3.3 能帶結構和態密度
基于DFT +U的計算方法, 對優化后的純銳鈦礦相TiO2和摻雜與缺陷體系的能帶結構和態密度進行了計算. 為方便比較, 各體系的能帶圖均選取費米能級在能量零點來觀察TiO2晶體沿布里淵區高對稱方向的能帶結構(如圖2所示). TiO2摻雜前后各體系對應的總態密度(total density of states, TDOS) 和分態密度 (partial density of states,PDOS)圖(如圖3所示)中的費米能級也取為能量零點.
由圖2(a)可知, 在高對稱點G處純銳鈦礦相TiO2幾乎為直接帶隙, 其帶隙寬度為 3.12 eV, 與實驗測量值3.23 eV十分接近. 對應的態密度圖3(a)顯示, 純 TiO2的價帶 (valence band, VB)和價帶頂 (valence band maximum, VBM)的態密度主要由 O 2p 軌道電子貢獻, 其導帶 (conduction band,CB)和導帶底 (conduction band minimum, CBM)的態密度則主要由Ti 3d軌道電子貢獻. 由此可知, 純銳鈦礦相TiO2的光吸收主要起源于電子在O 2p軌道與 Ti 3d軌道之間的躍遷. 另一方面,圖3(a)表明純TiO2的VB和CB中都含有O 2p和Ti 3d軌道電子, 這表明O原子與Ti原子之間具有部分共價鍵成分, 這一特性與文獻[16, 33]的報道一致.
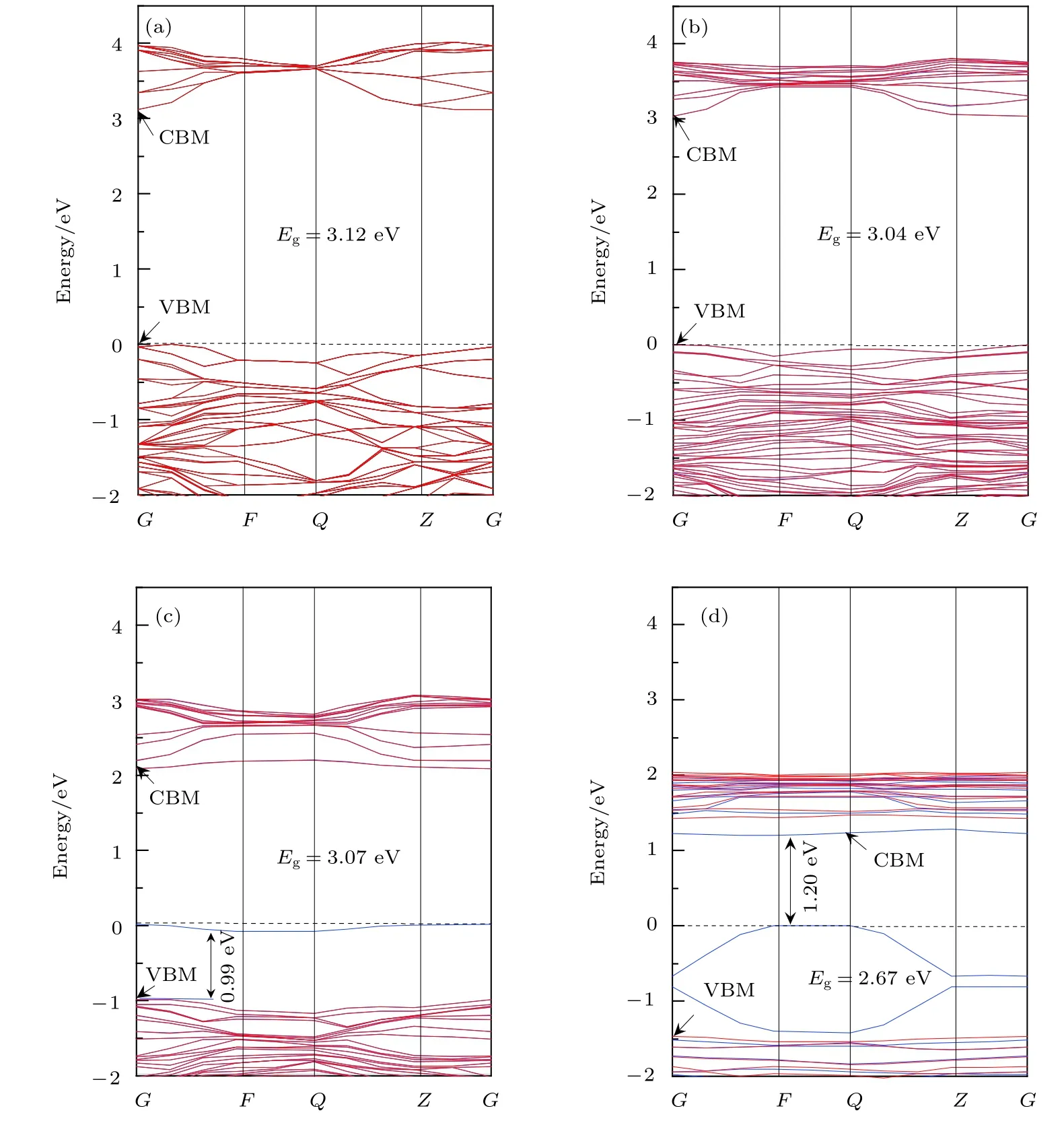
圖 2 純 TiO2 和 Ce/OV 摻 雜 TiO2 的 能 帶 結 構 圖 (圖 中 虛 線 代 表 費 米 能 級 EF) (a) Pure TiO2; (b) Ce-TiO2; (c) OV-TiO2;(d) Ce/OV-TiO2Fig.2. Band structure of anatase TiO2 before and after doping (the dashed lines represent the Fermi level, EF): (a) Pure TiO2;(b) Ce-doped TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2.
圖2(b)為Ce單摻雜TiO2的能帶圖. 由圖2(b)可知, 相對于純 TiO2而言, Ce-TiO2的 VBM 幾乎沒有移動, 而其CB整體朝低能方向發生了的移動, 從而導致Ce摻雜后TiO2的帶隙寬度窄化為3.04 eV. 結合圖 3(b)可知, 這種變化的主要原因可歸于 CBM 附近的 Ce 4f, Ce 5d, Ti 3d 及 O 2p軌道之間充分混合, 致使反鍵峰的位置相對于未摻雜前反鍵峰的位置明顯下移, 從而導致TiO2的CB下移、帶隙收縮. 顯然, 帶隙窄化可引起半導體的光吸收譜發生紅移. 與之相吻合是, 后文中計算所得的光吸收譜和相關理論和實驗研究[16,25,34]中均觀測到了這一紅移現象.
如圖2(c)所示, OV-TiO2的禁帶寬度相對于純TiO2發生了微小的收縮, 并且在費米能級附近出現了雜質能級. 態密度圖 3(c)顯示, O 2p, Ti 3d,Ti 3p, Ti 4s軌道之間充分疊加耦合, 在費米能級附近形成成鍵峰, 從而導致OV-TiO2的費米能級下方出現了具有占據態特性的受主能級. 這一結果與Zhang等[35]采用GGA +U方法計算得到的結果完全一致. 圖2(c)顯示該雜質能級與VBM相距0.99 eV, 這一能量差值與 Rumaiz 等[36]在實驗中觀測的結果極其相近. 他們觀測到含OV的TiO2帶隙中的雜質能級比VBM高1.0 eV. 此外, 對比圖3(c)與圖3(a)容易發現OV-TiO2的價帶寬度明顯發生了寬化. 這預示著電子具有更強的離域性, 十分有利于增強載流子的移遷能力.

圖 3 純 TiO2 和 Ce/OV 摻 雜 TiO2 的 態 密 度 圖 (圖 中 虛 線 代 表 費 米 能 級 EF) (a) Pure TiO2; (b) Ce-TiO2; (c) OV-TiO2;(d) Ce/OV-TiO2Fig.3. Comparison of partial density of states of anatase TiO2 before and after doping (the dashed lines represent the Fermi level,EF): (a) Pure TiO2; (b) Ce-doped TiO2; (c) OV-TiO2; (d) Ce/OV-TiO2.
比較圖 2(d)與圖 2(b)可知, Ce/OV共摻雜TiO2的禁帶寬度(2.67 eV)相對于Ce摻雜TiO2的禁帶寬度(3.04 eV)顯著窄化. 由此可見Ce摻雜TiO2中進一步引入OV, 可有效誘導Ce/OVTiO2的帶隙收縮. 態密度圖3(d)顯示Ce 4f和Ce 5d 進入 CBM, 并且與 Ti 3d 軌道充分混合, 致使反鍵峰所在位置明顯向低能方向移動, 從而導致Ce/OV-TiO2的導帶整體向低能方向大幅移動(如圖 3(b)所示); 同時, 在價帶區 Ce 5d, Ti 3p, Ti 4s與O 2p軌道電子之間的成鍵峰的位置均較單摻雜時的成鍵峰的位置向價帶低端能移動, 從而引起TiO2的價帶整體向低能明顯移動. 因此, Ce 與OV協同作用下可導致TiO2的帶隙顯著收縮. 另一方面, 能帶圖 2(d)顯示, Ce/OV-TiO2的禁帶中形成了極淺的受主能級. 這些雜質能級處于費米能級下方 (與 CBM相距 1.20 eV), 并且與 VBM充分交疊, 有利于電子從VBM躍遷至CBM. 這一效果與直接減小帶隙寬度相似, 極大地提高了TiO2對可見光的響應能力.
比較圖3(d)與圖3(b), 可觀測到相對于Ce-TiO2的價帶和導帶而言, Ce/OV-TiO2的價帶和導帶區的TDOS均向價帶低能端下移和延伸, 且峰形降低, 變得更為平滑和寬化. 這種價帶寬化與Ce 5d,Ti 4s與O 2p軌道電子在價帶低能區相互疊加引起電子的分布更為寬廣密切相關; 類似地, 圖3(c)中也觀測到價帶發生了寬化, 且其價帶寬化幅度略小于 Ce/OV-TiO2. 事實上, 這種價帶寬化現象, 是摻雜致使晶體的對稱性降低, 導致電子分布的非局域性增強而形成的. 此時, 態密度曲線的峰形降低、變得更為平滑和寬化[29,37]. 顯然, OV-TiO2和Ce/OV-TiO2中電子的非局域性的增強, 對提高光生載流子的遷移能力和TiO2的光催化活性十分有益.
3.4 帶邊位置
為探索摻雜前后半導體的導帶邊和價帶邊的還原氧化能力的變化, 可采用如下公式[38]:
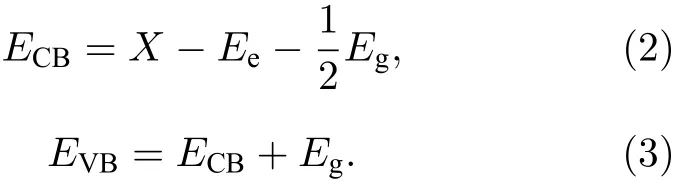
(2)和(3)式中, 導帶邊的還原電勢用ECB表示, 組成體系各原子的電負性的幾何平均值用X表示,自由電子的電勢值Ee以標準氫電極(normal hydrogen electrode, NHE)為參照 (~4.5 eV). 經計算純TiO2的導帶邊和價帶邊的電勢分別為?0.31 和 2.93 eV, 與實驗觀測值[39]極為相近. 圖 4為Ce/OV摻雜銳鈦礦相TiO2的能帶帶邊位置的示意圖.
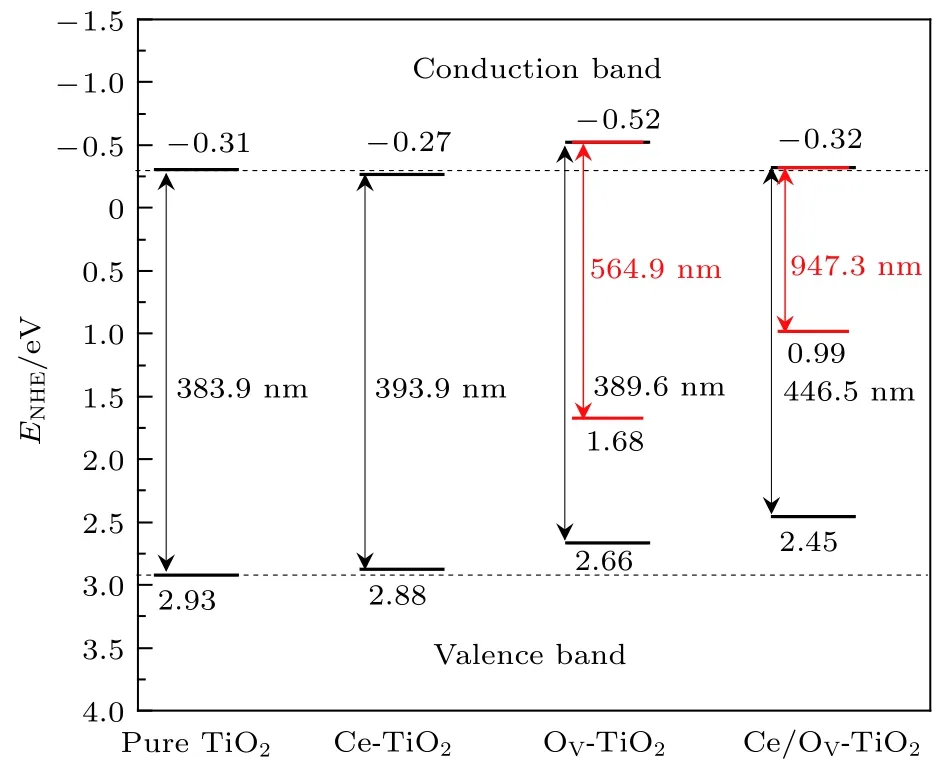
圖4 Ce/OV 摻雜銳鈦礦相 TiO2 的能帶帶邊位置示意圖(圖中各帶邊位置的電位均以NHE的電位為參考零點,粗黑線代表忽略雜質能級時的帶邊位置, 細紅線代表考慮雜質能級時的帶邊位置)Fig.4. Calculated band energy positions of pure TiO2, Ce-TiO2, OV-TiO2, Ce/OV-TiO2 (the energy scale is indicated by the normal hydrogen electrode in electron volts as a reference; the short thick lines and fine red lines represent the band energy positions of TiO2 by neglecting and considering the impurity levels, respectively).
如圖4所示, 相對于純TiO2的導帶邊和價帶邊的位置而言, Ce-TiO2(Ce 摻雜濃度為 2.08%)對應的帶邊位置分別向下和向上移動了0.035和0.047 eV. Ce-TiO2的帶邊位置移動方向與 Lenka等[25]報道的摻雜濃度為0.28%和3.0%的Ce/TiO2樣品的帶邊位置的移動方向一致. 但此兩種摻雜濃度的Ce/TiO2的帶邊移動幅度明顯大于本研究中Ce-TiO2的帶邊移動幅度. 究其原因除了與摻雜濃度不同這個因素有關外, 更重要的在于實驗制備中會形成CeO2, 因而可在TiO2的能帶中產生新的能級, 對半導體的帶邊電勢產生重要的影響. 顯然,Ce-TiO2的價帶邊的氧化能力因其價帶邊上移而有所削弱, 但2.88 eV的價帶邊電勢, 相對于氧化劑H2O2和O3的氧化電勢1.77和2.07 eV而言,可認為Ce-TiO2的價帶邊仍具有良好的氧化能力.另一方面, Ce-TiO2的導帶邊的還原能力卻因其導帶邊電勢位置下移, 而略有減弱. 值得指出的是,OV-TiO2的導帶帶邊顯著向上移動, 這表明TiO2中引入OV可明顯提高其導帶邊的光催化還原能力. Liu等[40]通過光致發光光譜(photoluminescence spectra, PL)檢測到 TiO2樣品中形成了 OV. 這些OV的存在可導致Ti4+向Ti3+轉化, 有利于降低光生電子-空穴對的復合[13]. 類似地, 圖 4 顯示, Ce/OV共摻雜TiO2的導帶邊的電勢位置略高于純TiO2和Ce單摻雜的導帶邊的電勢位置, 這說明其導帶邊的還原能力高于純TiO2和Ce單摻雜的情形.
根據光解水的基本條件[41]: 光催化劑的導帶邊的電勢應低于 H+/H2的還原電勢 (0 eV vs. NHE);另一方面, 光催化劑的價帶邊的電勢應高于O2/H2O的氧化電勢 (1.23 eV vs NHE). 結合圖 2 和圖 4 可知, Ce/OV-TiO2的基本帶隙明顯窄化, 對可見光具有良好的響應能力, 且其帶邊電勢可滿足上述光解水的基本要求. 因此, Ce/OV-TiO2可能是一種良好的適合于可見光光解水的光催化材料.
另一方面, 圖4顯示Ce/OV單(共)摻雜TiO2的光吸收閾值相對于純TiO2的光吸收閾值均向長波方向增長. 特別是, Ce/OV-TiO2的基本帶隙的光吸收響應明顯增長到可見光區域. 若考慮雜質能級的作用, OV-TiO2和Ce/OV-TiO2的光吸收能力分別拓展到可見光長波區域和紅外光區.
3.5 光吸收譜
半導體的光學性質的計算基本原理是基于直接帶間傳輸的介電函數的虛部公式[42]:
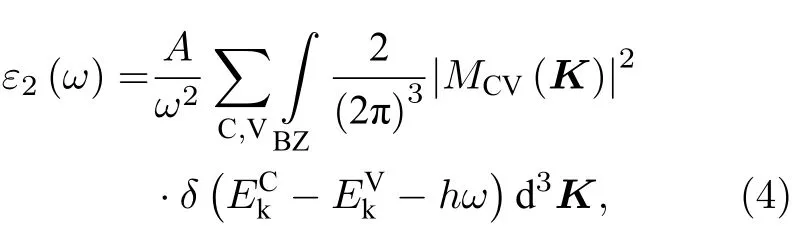
式中C, V, BZ和K分別表示導帶、價帶、第一布里淵區和倒格矢;|MCV(K)|2為動量躍遷矩陣元;A為與允許直接躍遷吸收系數有關的常數;ω為聲子頻率;EkC和EkV分別為導帶和價帶上的本征能級. 進一步由 Kramers-Kroning 變換關系[43], 可導出介電函數的實部ε1. 最終便可計算出材料的光吸收譜. 圖5為Ce/OV摻雜銳鈦礦TiO2的光吸收譜.
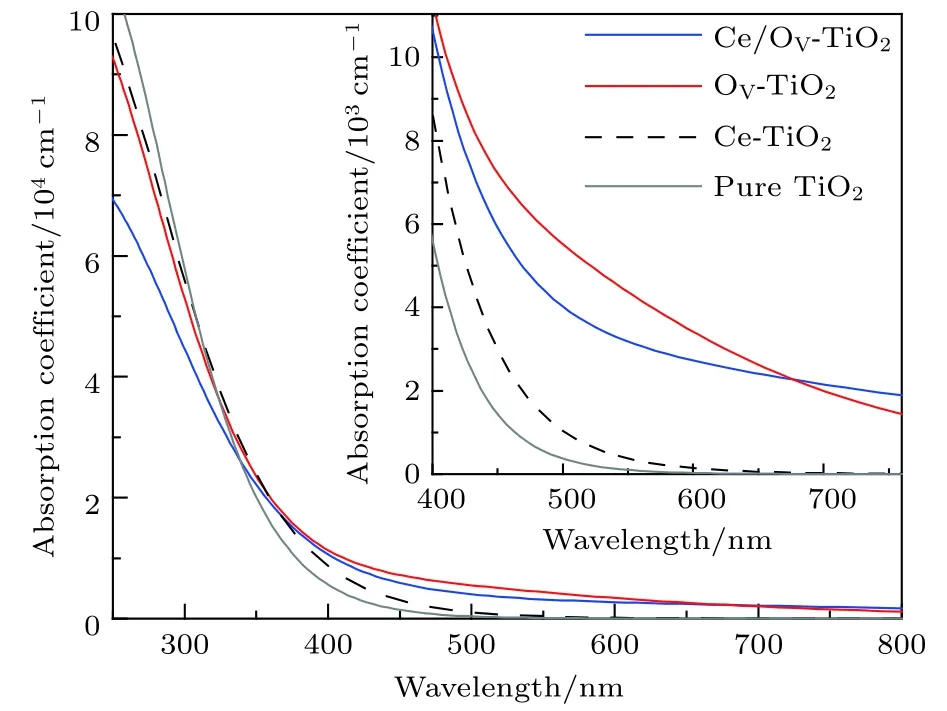
圖5 Ce/OV 摻雜銳鈦礦 TiO2 的光吸收譜 (插圖為可見光區光吸收譜的放大圖)Fig.5. Absorption spectra of pure and different doping TiO2 models (inset is the expanded absorption spectra in the visible region).
圖5顯示, 各摻雜體系的光吸收譜均發生了明顯的紅移, 且其在可見光區和更長波段區域的光吸收系數均高于純銳鈦礦相TiO2的光吸收系數. 其中, OV-TiO2和Ce/OV-TiO2在可見光區的吸收系數明顯大于Ce-TiO2的可見光光吸收系數, 且前者在大部分的可見光區的光吸收系數均高于后者.由此可見, Ce-TiO2中引入OV, 可有效增強TiO2對可見光的響應能力.
仔細比較Ce/OV-TiO2與OV-TiO2的光吸收譜, 可發現在 400.0—677.1 nm 之間, 前者比后者的光吸收系數略弱一些. 究其原因, 主要可歸因于如下兩個方面. 一是, 由圖3(c)和圖3(d)可發現,OV-TiO2與Ce/OV-TiO2在其費米能級附近的雜質能級上的電子態密度的大小存在較大差距, 前者明顯高于后者. 這預示著, 相對于Ce/OV-TiO2而言, OV-TiO2中雜質能級上的電子吸收光能向導帶躍遷的概率要大. 二是, 由圖 4 可知, Ce/OV-TiO2在可見光區域的光吸收閾值約為446.5 nm, 而OV-TiO2由于雜質能級的作用, 其光吸收閾值約為 564.9 nm. 這就是說, 前者對可見光的響應在一定區域內要弱于后者. 另一方面, 圖5顯示在波長大于 677.1 nm 的區域, Ce/OV-TiO2的光吸收系數略高于OV-TiO2的光吸收系數. 這一現象可以從圖2(d)和圖4得到解釋. 圖2(d)和圖4顯示,Ce/OV-TiO2的受主能級對更長波長的吸收響應能力顯著強于OV-TiO2中的雜質能級對更長波長的吸收響應能力, 因而導致在更長波段中Ce/OVTiO2的光吸收系數略高于OV-TiO2.
最近 , 許多實驗研究[25,34,44]報道 , Ce 摻雜TiO2可引起半導體的光吸收譜發生紅移, 并顯示出優良的光催化活性. 例如, Yan 等[44]實驗發現Ce摻雜濃度為1.0 mol%的Ce/TiO2的光吸收邊的紅移約為50.0 nm, 但摻雜濃度進一步增加時,TiO2的光吸收邊的紅移卻不再增加; 而Lenka等[25]在實驗中卻觀測到了Ce摻雜濃度增至10 mol%時, TiO2的光吸收邊的紅移約為 81.0 nm. 相比于Yan 等[44]和 Lenka 等[25]的實驗結果, 本文研究的Ce/OV-TiO2的光吸收邊的紅移約為 62.0 nm. 可見, 本研究的Ce/OV-TiO2的紅移幅度與實驗研究的Ce/TiO2的紅移幅度相近. 這是因為實驗制備的Ce摻雜TiO2不可避免地會產生一定濃度的OV, 本計算已考慮了這一因素, 因而計算結果與實驗相近; 若計算中不考慮OV的作用, 本研究發現Ce-TiO2的紅移幅度僅約為 10.0 nm. 另一方面,盡管本研究的Ce/OV-TiO2的紅移幅度與實驗結果相近, 但仍然存在明顯差距. 究其原因有: 實驗中 TiO2樣品的表面可形成 CeO2, 因而 OV和Ce的4f軌道在TiO2的能帶中形成了新的能級,從而促進了TiO2對可見光的響應能力. 此外, 實驗制備中因Ce離子氧化不充分, Ce4+和Ce3+可以同時存在, 從而形成氧化還原電對Ce3+/Ce4+[25,44],因而十分有利于提高光生電子-空穴對的遷移率和光量子產率[13]. 因此, 實驗中觀測到的Ce摻雜TiO2表現出了更優異的可見光光催化活性.
4 結 論
本文運用第一性原理LDA +U方法, 計算了Ce和OV單(共)摻雜銳鈦礦相 TiO2的晶格參數、缺陷形成能、電子結構和光吸收性質. 由計算結果可得到如下結論.
1) 在Ce/OV共摻雜TiO2的導帶區Ce 4f, Ce 5d,Ti 3d軌道之間充分混合, 致使反鍵峰的位置和導帶整體下移; 同時, 價帶區的 Ce 5d, Ti 3p, Ti 4s,Ti 3d, O 2p 軌道電子之間交疊耦合, 引起成鍵態峰位朝低能方向移動. 最終, 共摻雜TiO2的價帶明顯寬化, 在費米能級附近形成受主能級, 帶隙窄化為2.67 eV, 明顯小于純的和單摻雜TiO2的帶隙寬度. 因此, Ce/OV共摻雜可有效提高 TiO2對可見光的響應能力和增加其光吸收范圍.
2) Ce/OV共摻雜使 TiO2的晶格畸變, 并在其八面體單元中誘生出顯著的電偶極矩, 從而可有效提高光生電子-空穴對的遷移能力和半導體的光催化活性; 另一方面, Ce摻雜 TiO2中引入 OV可增強導帶邊的還原能力, 共摻雜后TiO2的能帶邊的電勢位置滿足光解水的基本條件, 且在可見光區的光吸收強度明顯高于純TiO2和Ce單摻雜TiO2.
綜上所述, Ce/OV共摻雜TiO2的能帶結構可滿足可見光光解水的基本要求. 本文的研究為相關理論和實驗的進一步研究提供了良好的理論基礎.
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
